 2021, Vol. 32
2021, Vol. 32 


b Department of Chemistry, Clark Atlanta University, Atlanta, Georgia 30314, United States
Covalent organic frameworks (COFs), a new class of frontier crystalline porous materials constructed by linking organic building blocks through reversible covalent bonds, have garnered enormous attention owing to their enticing structural uniqueness such as precise skeleton periodicity, ultralow density, high porosity and modular pore metrics [1-4], which underpin their widespread potential applications in gas absorption [5-7], heterogeneous catalysis [8, 9], chemical sensing [10-12], lithium-ion batteries [13, 14], chromatographic separation [15-18], optoelectronics [19-21], etc. Despite a vast number of multifunctional COFs reported to date, the construction of chiral COFs is rather scarce and remains a formidable challenge, largely due to the intrinsic conflict between crystallinity and asymmetry [1, 22]. Furthermore, developing chiral COFs by utilizing enantiopure building blocks might result in unwanted achiral polymers owing to the linker racemization under harsh solvothermal conditions, as being demonstrated in the synthesis of chiral metal-organic frameworks (MOFs) [23-25].
To impart chirality in COFs, two main strategies, i.e., post-synthetic modification [26, 27], and de novo approach [15-18, 28, 29] have been proved as effective means to integrate chiral functionalities into two- or three-dimensional (2D or 3D) COFs. For instance, in 2015, Jiang and co-workers developed the robust chiral COF by a post-synthetic modification strategy, wherein the chiral moiety of (S)-pyrrolidine was introduced into the pore wall of an achiral COF via click reaction [27]. Later in 2016, Yan's group reported a de novo synthesis of chiral COFs by utilizing an enantiopure monomer that was functionalized with (+)-diacetyl-L-tartaric anhydride [15]. In 2017, Cui's group developed two chiral Zn(salen)-based COFs through Schiff-base reaction between enantiopure 1, 2-diaminocyclohexane and trisalicylaldehydes [29]. Despite significant progress in the design and synthesis of chiral COFs, the underlying mechanistic study of chiral COFs formation remains substantially elusive [30]. On the other hand, achiral COFs bearing carboxyl groups have been synthesized through a facile post-synthetic modification of hydroxyl COFs [31, 32]. However, the construction of carboxyl-functionalized chiral COFs has never been realized.
Herein, we report a de novo synthesis of an enantiomeric pair of 2D hydrazone-linked chiral COFs bearing pre-installed hydroxyl groups, termed (S)-HthBta-OH and (R)-HthBta-OH COFs (Scheme 1), via the Schiff-base reaction of enantiopure 2, 5-bis(2-hydroxypropoxy)terephthalohydrazide ((S)-Hth and (R)-Hth) with 1, 3, 5-benzenetricarboxaldehyde (Bta). The formation mechanism of such hydroxyl-functionalized chiral COFs was systematically investigated by means of rigorous kinetic studies. Remarkably, time-dependent VCD spectra uncovered an inverted chiral signal from the positive Cotton effect of (S)-Hth monomer to the negative Cotton effect of (S)-HthBta-OH COF. To the best of our knowledge, this represents the first report of VCD chiral signal inversion in chiral COFs. Moreover, post-synthetic modification allowed for the reaction between hydroxyl groups of (S)- and (R)-HthBta-OH COFs and succinic anhydride, leading to the first construction of carboxyl-functionalized (S)- and (R)-HthBta-COOH COFs, respectively (Scheme 1). Furthermore, the homochirality and crystallinity of (S)- and (R)-HthBta-COOH COFs remained intact during the post-synthetic modification process owing to the chemical robustness of pristine hydrazone-linked chiral COFs.

|
Download:
|
| Scheme 1. Synthesis of chiral (S)- and (R)-HthBta-OH COFs as well as the corresponding (S)- and (R)-HthBta-COOH COFs by post-synthetic modification. | |
{kind=link}
(S)- and (R)-HthBta-OH COFs could be synthesized by the Schiff-base condensation between Bta (3.2 mg, 0.02 mmol) and enantiopure (S)-Hth or (R)-Hth (10.3 mg, 0.03 mmol) in a degassed mixture of 1, 4-dioxane (0.5 mL), mesitylene (0.5 mL) and 6 mol/L aqueous acetic acid (0.1 mL) at 110 ℃ for 3 days, giving rise to pale-yellow solids in excellent yields (∼90%). The resulting chiral COFs were found to be insoluble in common organic solvents such as acetone, ethanol, tetrahydrofuran, dichloromethane, and N, N-dimethylformamide (DMF). Powder X-ray diffraction (PXRD) was employed to assess the crystallinity of (S)- and (R)-HthBta-OH COFs. (S)-HthBta-OH COF exhibited a strong PXRD peak at 3.4°, and two minor peaks at 6.9° and 26.4° (Fig. 1a), corresponding to the (100), (200), and (001) facets, respectively. Furthermore, (S)- and (R)-HthBta-OH COFs showed identical PXRD patterns (Fig. S1a in Supporting information), indicating the long-range ordered and isostructural structures of both two COFs. To elucidate the crystalline structures of (S)- and (R)-HthBta-OH COFs, two possible 2D layered chiral structures, that is, eclipsed AA stacking with a space group of P6 (Fig. S1e, Tables S1 and S3 in Supporting information) and staggered AB stacking with a space group of P63 (Fig. S1f, Tables S2 and S4 in Supporting information), were simulated by using Materials Studio program [33]. The experimental PXRD patterns of (S)- and (R)-HthBta-OH COFs reproduced well with the simulated patterns based on the eclipsed AA packing mode with respect to peak position and intensity (Fig. 1b and Fig. S1b in Supporting information) but did not match well with those generated from the staggered AB packing of 2D layers (Fig. 1c and Fig. S1c in Supporting information). Pawley refinements based on the eclipsed AA stacking structures were further conducted and gave rise to the optimized parameters of unit cell: a = b = 30.32 Å, c = 4.08 Å, α = β = 90°, γ = 120° for (S)-HthBta-OH COF, and a = b = 30.33 Å, c = 4.07 Å, α = β = 90°, γ = 120° for (R)-HthBta-OH COF, with good agreement factors (Rwp = 2.80%, Rp = 2.16% for (S)-HthBta-OH COF and Rwp = 6.25%, Rp = 4.50% for (R)-HthBta-OH COF).

|
Download:
|
| Fig. 1. (a) Experimental (red) and Pawley refined (black) PXRD patterns of (S)-HthBta-OH COF. (b) Simulated PXRD pattern for the eclipsed AA mode. (c) Simulated PXRD pattern for the staggered AB mode. (d) The difference between the experimental and the refined PXRD patterns. (e) Eclipsed AA packing and (f) staggered AB packing of (S)-HthBta-OH COF. | |
{kind=link}
Considering (S)- and (R)-HthBta-OH COFs were enantiomers, we selected (S)-HthBta-OH COF as a representative example for thorough structural characterization by various analytical methods. Elemental analysis of (S)-HthBta-OH COF revealed that the experimental C, N and H contents corroborated well with the theoretical values (Calcd. for C10H11N2O3 (%): C, 57.97; H, 5.35; N, 13.52. Found: C, 51.70; H, 5.82; N, 11.38). The Fourier transform infrared (FT-IR) spectrum of (S)-HthBta-OH COF (Fig. 2a) revealed two characteristic vibrational bands at 1618 and 1229 cm−1, which were assigned to the C=N bonds. Furthermore, the stretching bands arising from amine (3100–3400 cm−1) and aldehyde (1699 cm−1) largely decreased or even disappeared in comparison to those of (S)-Hth and Bta, further verifying the effective condensation reaction between COF precursors. The 13C cross-polarization magic angle spinning (CP-MAS) solid-state NMR spectrum of (S)-HthBta-OH COF showed an intense signal at 150 ppm, corresponding to the carbons in C=N bonds, while the aldehyde carbon signal was barely observed (Fig. 2b). Thermogravimetric analysis (TGA) demonstrated that (S)-HthBta-OH COF was stable up to 320 ℃ under the air atmosphere (Fig. 2c). The scanning electron microscopy (SEM) showed that (S)-HthBta-OH COF adopted a unique hollow rod-shaped morphology with the length being a few microns (Fig. 2d). The porosity of (S)-HthBta-OH COF was evaluated by nitrogen adsorption-desorption isotherms at 77 K, which exhibited a characteristic Type Ⅰ shape, indicating the micro-porosity of the obtained COF material. The Brunauer-Emmett-Teller (BET) surface area of (S)-HthBta-OH COF was calculated to be 794 m2/g (Fig. 2e), which was higher than the reported hydrazone-linked achiral 2D COFs [12, 34]. The total pore volume was determined to be 0.35 cm3/g on the basis of a single point measurement at P/Po = 0.99. Furthermore, the pore width of (S)-HthBta-OH COF estimated using nonlocal density functional theory (NLDFT) was 1.5 nm (Fig. 2f), which was in good agreement with the predicted value for an AA stacking mode (1.6 nm).

|
Download:
|
| Fig. 2. (a) FT-IR spectra of (S)-Hth, Bta and (S)-HthBta-OH COF. (b) Solid-state 13C CP-MAS NMR spectrum of (S)-HthBta-OH COF. (c) TGA curve of (S)-HthBta-OH COF under the air atmosphere. (d) SEM image of (S)-HthBta-OH COF. (e) N2 adsorption (●) and desorption (○) isotherm of (S)-HthBta-OH COF and (f) pore size distribution of (S)-HthBta-OH COF. | |
{kind=link}
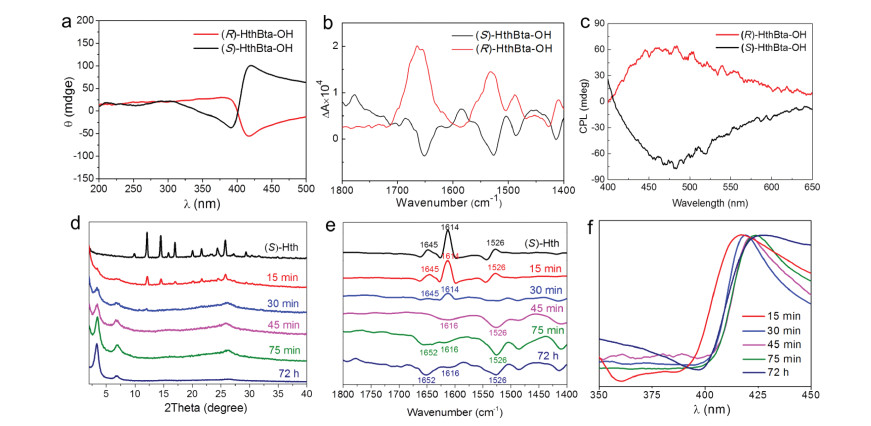
To assess the chirality of Hth monomers and the resulting hydroxyl-functionalized COFs, electronic circular dichroism (ECD) spectroscopy was applied in the solid-state at room temperature. As depicted in Fig. 3a, ECD spectra of (S)- and (R)-HthBta-OH COFs clearly showed mirror images of each other, indicating that they were a pair of enantiomers. In the wavelength range of 350−450 nm, (S)-HthBta-OH COF displayed a positive Cotton effect at 427 nm and negative dichroic signal at 397 nm, whereas (R)-HthBta-OH COF showed an enantiomeric ECD pattern at the similar wavelengths. The ECD spectra of (S)- and (R)-Hth monomers also affirmed their enantiomeric nature, in which opposite Cotton effects at the same wavelengths (main peaks at ∼285, 314 and 356 nm) were observed (Fig. S2a in Supporting information). Since vibrational circular dichroism (VCD) spectroscopy could extend the range of ECD measurements into the infrared region, it has been widely used as a potent analytical tool for probing the structure of chiral porous frameworks [35, 36]. As depicted in Fig. 3b and Fig. S2b (Supporting information), the VCD spectra of both (S)/(R)-Hth monomers and (S)/(R)-HthBta-OH COFs, which corroborated well with the IR spectra (Fig. S2b), exhibited satisfactory mirror images of each other, offering another evidence of their enantiomeric nature. Moreover, (S)- and (R)-HthBta-OH COFs displayed strong fluorescence emission in the solid-state (Fig. S3 in Supporting information), which inspired us to explore their circularly polarized luminescence (CPL) behaviors. As illustrated in Fig. 3c, (S)- and (R)-HthBta-OH COFs also exhibited good mirror-image CPL signals centered at 480 nm. Taken together, the combined results of ECD, VCD, and CPL demonstrated the successful transformation of chirality from chiral Hth moieties to HthBta-OH COFs.

|
Download:
|
| Fig. 3. (a) ECD, (b) VCD and (c) CPL spectra of the (S)- and (R)-HthBta-OH COFs. (d) PXRD patterns, (e) VCD and (f) ECD spectra of the (S)-HthBta-OH COF products obtained at different reaction times ranging from 15 min to 72 h, respectively. | |
{kind=link}
It is worth noting that the signals in the VCD spectra of (S)- and (R)-HthBta-OH COFs were inverted compared to those of chiral Hth monomers, which intrigued us to further elucidate the formation process of (S)- and (R)-HthBta-OH COFs. Toward this end, the synthesis of (S)-HthBta-OH COF as a representative example was undertaken in the identical solvothermal conditions with varied reaction times. The products were collected by filtration, washed with 1, 4-dioxane and THF, and then subjected to PXRD, VCD, and ECD measurements. As shown in Fig. 3d, the PXRD pattern of the product obtained in 15 min exhibited a set of diffraction peaks arising from the (S)-Hth monomer and two very weak peaks in the smaller angle region, indicating the incompleteness of Schiff-base reaction and the amorphous nature of (S)-HthBta-OH COF. When the reaction time was extended to 30 min, the peak intensity of (S)-Hth significantly decreased, while those of (S)-HthBta-OH COF increased. Notably, the (S)-Hth monomer completely disappeared after 45 min, whereas medium-strong diffraction peaks at 3.5°, 7.0° and 26.4° were observed, indicative of an amorphous-crystalline transformation in (S)-HthBta-OH COF. The crystallinity of (S)-HthBta-OH COF further increased in 75 min, which was comparable to that obtained in 72 h, revealing the rapid formation of (S)-HthBta-OH COF, which was drastically faster than the conventional hydrazone-linked COFs [12]. To gain deeper insights into the chiral signal change from monomers to chiral COFs, we conducted the time-dependent VCD measurements. As illustrated in Fig. 3e, the VCD spectrum of (S)-Hth exhibited three intense positive peaks at 1645, 1614 and 1526 cm−1, which were ascribed to the C=O stretching vibration, N-H bending vibration of -NH2 group, and the skeleton vibration of the aromatic ring, respectively. The intensities of the three positive peaks continuously decreased as the reaction times were kept at 15 and 30 min. These three positive peaks disappeared after 45 min, whereas new negative peaks at 1616, 1526 cm−1 arose, indicating the complete consumption of (S)-Hth and the formation of (S)-HthBta-OH COF. In particular, the VCD spectrum of (S)-HthBta-OH COF obtained in 75 min (similar to the one synthesized in 72 h) displayed three obvious negative peaks at 1652, 1616, 1526 cm−1, corresponding to the C=O stretching vibration, the C=N stretching vibration, and the aromatic ring skeleton vibration of the resulting chiral COF. These VCD results were in line with the PXRD and IR analyses (Fig. S4 in Supporting information). Remarkably, the VCD spectra revealed a unique VCD chiral signal inversion from the positive Cotton effect of (S)-Hth monomer to the negative Cotton effect of (S)-HthBta-OH COF, which has never been documented in chiral COFs. Given that the VCD chiral signals could be profoundly affected by the conformations of chiral compounds [37, 38], we postulated that such a rare chiral signal inversion from monomer to COF could be attributed to the conformation change of (S)-Hth moieties in the resulting (S)-HthBta-OH COF. While the conformation change of (S)-Hth moieties was due to the π-π stacking and hydrogen bonding interactions between the adjacent COF layers. Moreover, the time-dependent synthesis of (S)-HthBta-OH COF was monitored using ECD spectroscopy (Fig. 3f). The ECD spectra of the products obtained from 15 min to 72 h presented strong positive peaks centered at 416, 419, 421, 424, 426 nm, respectively. Such bathochromic shifts could be due to the formation of the more extended π-systems in (S)-HthBta-OH COF.
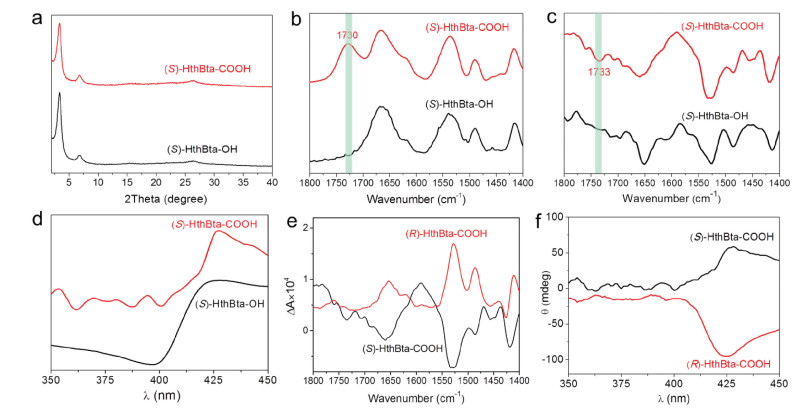
On account of the high crystallinity, large surface area, good stability, and abundant free hydroxyl functionalities in the homochiral COFs, we rationally transformed (S)- and (R)-HthBta-OH COFs into carboxyl-functionalized chiral COFs through the post-synthetic modification. Specifically, an enantiomeric pair of chiral COFs bearing carboxyl groups, namely (S)- and (R)-HthBta-COOH COFs, were synthesized by a facile reaction between the hydroxyl groups on the backbones of (S)- and (R)-HthBta-OH COFs and succinic anhydride in DMF at 80 ℃ for 4 days (Scheme 1). The resulting carboxyl-functionalized chiral COFs were thoroughly characterized by a number of analytical techniques. The PXRD pattern of (S)-HthBta-COOH COF displayed an intense diffraction peak at 3.4° and two weak peaks at 6.8° and 26.4°, which matched well with those of the parent (S)-HthBta-OH COF (Fig. 4a), indicating the packing mode of (S)-HthBta-COOH COF did not change after the post-synthetic modification of (S)-HthBta-OH COF. SEM images of (S)-HthBta-COOH COF displayed similar morphology to that of (S)-HthBta-OH COF (Fig. S5 in Supporting information), further confirming no obvious morphological and structural damage occurred during the modification process. TGA revealed that (S)-HthBta-COOH COF possessed comparable thermal stability (325 ℃) to the pristine (S)-HthBta-OH COF (Fig. S6 in Supporting information). The BET surface area and pore size of (S)-HthBta-COOH COF were determined to be 223 m2/g (Fig. S7 in Supporting information) and 1.2 nm (Fig. S8 in Supporting information), respectively, which were smaller than those of the (S)-HthBta-OH COF owing to the successful grafting of bulky carboxyl groups onto the pore wall of the pristine COFs.

|
Download:
|
| Fig. 4. (a) PXRD patterns, (b) FT-IR spectra, (c) VCD spectra and (d) ECD spectra of (S)-HthBta-OH and (S)-HthBta-COOH COFs. (e) VCD and ECD spectra of (S)- and (R)-HthBta-COOH COFs. | |
{kind=link}
The FT-IR spectrum of (S)-HthBta-COOH COF showed an emerging peak at 1730 cm−1, corresponding to the stretching vibration of C=O in the newly formed ester and free carboxylic acid, suggesting the effective ring-opening reaction between succinic anhydride and the hydroxyl groups of (S)-HthBta-OH COF (Fig. 4b). The VCD spectrum of (S)-HthBta-COOH COF presented the same trend to that of the pristine (S)-HthBta-OH COF (Fig. 4c), where a set of negative peaks at similar wavenumbers were observed, indicating the preserved chirality of COFs and intact conformations of (S)-Hth moieties in (S)-HthBta-COOH COF. It is worth noting that a new negative peak at 1733 cm−1 was also found in the VCD spectrum of (S)-HthBta-COOH COF, further validating the formation of the ester and carboxylic acid groups. Furthermore, the ECD spectrum of (S)-HthBta-COOH COF resembled that of (S)-HthBta-OH COF, wherein a positive peak at the wavelength of 427 nm was observed (Fig. 4d). More importantly, both VCD and ECD spectra of (S)- and (R)-HthBta-COOH COFs were mirrored images of each other, confirming their enantiomeric nature (Figs. 4e and f). The successful construction of carboxyl-functionalized chiral COFs without linker racemization under harsh modification conditions could be attributed to the exceptional chemical robustness of the hydroxyl-functionalized hydrazone-linked COFs. No reversal of the chiral signals was observed in both VCD and ECD spectra of (S)-HthBta-COOH COF compared with those of (S)-HthBta-OH COF, illustrating the retained conformation of the (S)-Hth moieties in the resulting (S)-HthBta-COOH COF.
In summary, we have developed a de novo synthesis of a pair of 2D hydrazone-linked (S)- and (R)-HthBta-OH chiral COFs with pre-installed hydroxyl functionalities. VCD, ECD and CPL measurements unambiguously proved that (S)- and (R)-HthBta-OH COFs were enantiomers. Moreover, the formation process of hydroxyl-functionalized chiral COFs was monitored using rigorous time-dependent PXRD, VCD and ECD studies. Remarkably, the VCD measurement showed an unprecedented chiral signal inversion from the positive Cotton effect of (S)-Hth monomer to the negative Cotton effect of (S)-HthBta-OH COF, which was ascribed to the possible conformation change of (S)-Hth moieties in (S)-HthBta-OH COF. Thanks to their excellent crystallinity, good chemical stability, high porosity, and abundant hydroxyl functionalities, we post-synthetically converted robust (S)- and (R)-HthBta-OH COFs into carboxyl-functionalized (S)- and (R)-HthBta-COOH COFs with the retention of homochirality and crystallinity, indicating no linker racemization and structural damage during the post-synthetic modification process. This work not only paves a new avenue to the construction of homochiral COFs possessing distinct functionalities but also sheds a deeper light on how the chiral signals changed from chiral monomers to homochiral COFs. We envision that the resulting stable hydrazone-linked homochiral COFs with abundant hydroxyl and carboxyl functional groups may be employed as good candidates for chiral chromatographic separation and asymmetric catalysis. These potential applications are currently under study in our group.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (Nos. 21603076 and 21571070) and the Natural Science Foundation of Guangdong Province (No. 2018A030313193).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.063.
| [1] |
K. Geng, T. He, R. Liu, et al., Chem. Rev. 120 (2020) 8814-8933. DOI:10.1021/acs.chemrev.9b00550 |
| [2] |
C.S. Diercks, O.M. Yaghi, Science 355 (2017) 923. |
| [3] |
S.L. Cai, W.G. Zhang, R.N. Zuckermann, et al., Adv. Mater. 27 (2015) 5762-5770. DOI:10.1002/adma.201500124 |
| [4] |
S.Y. Ding, W. Wang, Chem. Soc. Rev. 42 (2013) 548-568. DOI:10.1039/C2CS35072F |
| [5] |
S.S. Han, H. Furukawa, O.M. Yaghi, et al., J. Am. Chem. Soc. 130 (2008) 11580-11581. DOI:10.1021/ja803247y |
| [6] |
Z. Xiang, R. Mercado, J.M. Huck, et al., J. Am. Chem. Soc. 137 (2015) 13301-13307. DOI:10.1021/jacs.5b06266 |
| [7] |
S.L. Cai, K. Zhang, J.B. Tan, et al., ACS Macro Lett. 5 (2016) 1348-1352. DOI:10.1021/acsmacrolett.6b00805 |
| [8] |
S.Y. Ding, J. Gao, Q. Wang, et al., J. Am. Chem. Soc. 133 (2011) 19816-19822. DOI:10.1021/ja206846p |
| [9] |
H. Li, Q. Pan, Y. Ma, et al., J. Am. Chem. Soc. 138 (2016) 14783-14788. DOI:10.1021/jacs.6b09563 |
| [10] |
S. Dalapati, S. Jin, J. Gao, et al., J. Am. Chem. Soc. 135 (2013) 17310-17313. DOI:10.1021/ja4103293 |
| [11] |
S.Y. Ding, M. Dong, Y.W. Wang, et al., J. Am. Chem. Soc. 138 (2016) 3031-3037. DOI:10.1021/jacs.5b10754 |
| [12] |
G. Chen, H.H. Lan, S.L. Cai, et al., ACS Appl. Mater. Interfaces 11 (2019) 12830-12837. DOI:10.1021/acsami.9b02640 |
| [13] |
J. Wang, L. Si, Q. Wei, et al., ACS Appl. Nano Mater. 1 (2017) 132-138. |
| [14] |
X. Li, H. Wang, H. Chen, et al., Chem 6 (2020) 933-944. DOI:10.1016/j.chempr.2020.01.011 |
| [15] |
H.L. Qian, C.X. Yang, X.P. Yan, Nat. Commun. 7 (2016) 12104. DOI:10.1038/ncomms12104 |
| [16] |
K. Zhang, S.L. Cai, Y.L. Yan, et al., J. Chromatogr. A 1519 (2017) 100-109. DOI:10.1016/j.chroma.2017.09.007 |
| [17] |
X. Han, J. Huang, C. Yuan, et al., J. Am. Chem. Soc. 140 (2018) 892-895. DOI:10.1021/jacs.7b12110 |
| [18] |
X.L. Huang, H.H. Lan, Y.L. Yan, et al., Sep. Sci. Plus 2 (2019) 120-128. DOI:10.1002/sscp.201800146 |
| [19] |
J.W. Colson, A.R. Woll, A. Mukherjee, et al., Science 332 (2011) 228-231. DOI:10.1126/science.1202747 |
| [20] |
M. Dogru, M. Handloser, F. Auras, et al., Angew. Chem. Int. Ed. 52 (2013) 2920-2924. DOI:10.1002/anie.201208514 |
| [21] |
S.L. Cai, Y.B. Zhang, A.B. Pun, et al., Chem. Sci. 5 (2014) 4693-4700. DOI:10.1039/C4SC02593H |
| [22] |
G. Liu, J. Sheng, Y. Zhao, Sci. China Chem. 60 (2017) 1015-1022. |
| [23] |
L. Bu, X.H. Chen, Chem. Mater. 18 (2006) 1857-1860. DOI:10.1021/cm052801c |
| [24] |
R. Vaidhyanathan, D. Bradshaw, J.N. Rebilly, et al., Angew. Chem. Int. Ed. 45 (2006) 6495-6499. DOI:10.1002/anie.200602242 |
| [25] |
R.E. Morris, X.H. Bu, Nat. Chem. 2 (2010) 353-361. DOI:10.1038/nchem.628 |
| [26] |
H. Xu, J. Gao, D. Jiang, Nat. Chem. 7 (2015) 905-912. DOI:10.1038/nchem.2352 |
| [27] |
H. Xu, X. Chen, J. Gao, et al., Chem. Commun. 50 (2014) 1292-1294. DOI:10.1039/C3CC48813F |
| [28] |
H.S. Xu, S.Y. Ding, W.K. An, et al., J. Am. Chem. Soc. 138 (2016) 11489-11492. DOI:10.1021/jacs.6b07516 |
| [29] |
X. Han, Q. Xia, J. Huang, et al., J. Am. Chem. Soc. 139 (2017) 8693-8697. DOI:10.1021/jacs.7b04008 |
| [30] |
X. Han, J. Zhang, J. Huang, et al., Nat. Commun. 9 (2018) 1294. DOI:10.1038/s41467-018-03689-9 |
| [31] |
Q. Lu, Y. Ma, H. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 6042-6048. DOI:10.1002/anie.201712246 |
| [32] |
N. Huang, X. Chen, R. Krishna, et al., Angew. Chem. Int. Ed. 54 (2015) 2986-2990. DOI:10.1002/anie.201411262 |
| [33] |
Accelrys Software Inc, Materials Studio 5.0: Modeling Simulation for Chemical and Material San Diego, CA, 2009.
|
| [34] |
F.J. Uribe-Romo, C.J. Doonan, H. Furukawa, et al., J. Am. Chem. Soc. 133 (2011) 11478-11481. DOI:10.1021/ja204728y |
| [35] |
I. Burneo, K.C. Stylianou, I. Imaz, D. Maspoch, Chem. Commun. 50 (2014) 13829-13832. DOI:10.1039/C4CC06190J |
| [36] |
S.Y. Zhang, D. Li, D. Guo, et al., J. Am. Chem. Soc. 137 (2015) 15406-15409. DOI:10.1021/jacs.5b11150 |
| [37] |
E. Tur, G. Vives, G. Rapenne, et al., Tetrahedron Asymmetry 18 (2007) 1911-1917. DOI:10.1016/j.tetasy.2007.07.020 |
| [38] |
Z. Dezhahang, M.R. Poopari, J. Cheramy, Y.J. Xu, Inorg. Chem. 54 (2015) 4539-4549. DOI:10.1021/acs.inorgchem.5b00386 |