 2021, Vol. 32
2021, Vol. 32 


Because of structural complexities and significant activities, diverse diterpenoid alkaloids have elicited continuous interests from chemical and pharmacological communities over a century [1]. Around 1300 diterpenoid alkaloid natural products have be reported mainly from ranunculaceous plants of the Aconitum genus [2]. The lateral root of Aconitum carmichaelii Debx., generally designated as "Fu Zi" (Chinese), is a popular Chinese herbal medicine for the treatment of various pains including rheumatalgia, neuralgia, and angina [3]. As fatal toxicity, this plant medicine must very cautiously be utilized after long-time decocting or properly processing to remove toxicity [3]. Previous studies showed that poisonous aconitane-alkaloidal diesters could be decomposed into less or no poisonous alkaloidal alcohols or monoesters [3]. However, the aconitane-alkaloidal alcohols and monoesters had relatively low analgesic activity when compared with that of the diesters as well as the decocting extract [3]. This indicates that the extract contains unknown active constituents. Although over 120 constituents have been reported from A. carmichaelii, organic solvents (EtOH or MeOH) as well as acid (HCl) and base (NH4OH) were applied to preparation and fractionation of the crude extracts [3c, 4]. Hence, to be relatively close to the clinical application protocol, we extracted the drug materials with water in our program to reinvestigate the chemical constituents of traditional Chinese medicines, mainly focusing on minor compounds [5]. From an aqueous extract of "Fu Zi", we have reported 52 diterpenoid alkaloids including six unprecedented sulfonated C20-diterpenoid alkaloids with two novel skeletons deriving from semipinacol rearrangements of napelline- and atisane-types of structural architectures, together with other structural diverse compounds [6]. A continued investigation of remaining active fractions from the extract has led to characterization of further two C20-diterpenoid alkaloids with different novel carbon skeletons originating from the Criegee rearrangements, designated as aconapelsulfonines A (1) and B (2), respectively (Fig. 1). Herein, we describe their isolation (Supporting information), structural elucidation, possible biogenetic routes, and pharmacological activities.

|
Download:
|
| Fig. 1. Structures of 1 and 2. | |
Compound 1, colorless prisms (MeOH-H2O, 3:1; m.p. > 300 ℃), [α]D20 -11.9 (c 0.21, H2O), showed a strong broad IR absorption (3200-3500 cm-1) for hydroxy functionalities. The nitrogen- and sulfur-containing molecular formula of 1 was determined as C22H33NO8S by positive and negative mode HR-ESIMS at m/z 472.1992 [M+H]+ (calcd. for C22H34NO8S, 472.2000) and 470.1848 [M-H]- (calcd. for C22H32NO8S, 470.1854), together with NMR spectroscopic data (Table 1). The 1H NMR spectroscopic data of 1 in D2O revealed the presence of following structures units: (a) a quaternary methyl [δH 0.88 (s, H3-18)]; (b) an N-ethyl [δH 3.24 (2H, m, H2-21) and 1.35 (3H, t, J = 7.2Hz, H3-22)]; (c) two sulfur- and nitrogen-bearing methylenes attached to quaternary carbons [δH 3.71 and 3.22 (each d, J = 14.4Hz, H-17a and H-17b) and 3.32 and 2.92 (each d, J = 13.8Hz, H-19a and H-19b)]; (d) four oxygen- and nitrogen-bearing methines [δH 4.12 (dd, J = 10.2 and 7.2, H-1), 4.32 (brs, H-13), 4.22 (brs, H-15), and 3.95 (brs, H-20)]; (e) three aliphatic methines [δH 1.69 (d, J = 8.4Hz, H-5), 2.59 (d, J = 3.6Hz, H-7), and 2.38 (dd, J = 10.8 and 8.4Hz, H-9)]; and (f) five aliphatic methylenes [δH 1.46 ― 3.10 (10 H, partially overlapped multiplets, H2-2, H2-3, H2-6, H2-11, and H2-14). The presence of the above structural units was confirmed by further analysis of the 13C NMR, DEPT, and HSQC experimental data, which led to unambiguous assignments of resonances in the NMR spectra of 1, including five quaternary carbons resonated at δC 35.4 (C-4), 43.2 (C-8), 51.7 (C-10), 103.2 (C-12) and 108.9 (C-16). The aforementioned spectroscopic data suggested that 1 was a sulfonated unusual C20-diterpenoid alkaloid [6g, 6i, 6j]. Especially the chemical shift values of the quaternary carbons C-12 and C-16 indicated that there were semi-acetal and acetal functionalities in the structure of 1, which were highly unusual in the C20-diterpenoid alkaloids. Subsequent comprehensive explanation of correlation signals in the 1H-1H COSY and HMBC spectra of 1 constructed an unique skeletal structure.
|
|
Table 1 NMR spectroscopic data for 1 and 2 in D2O.a |

{kind=link}
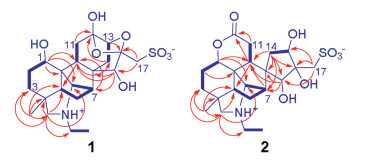
Four structural fragments in 1 (Fig. 2, bold lines) were elucidated from the homonuclear vicinal coupling cross-peaks of H-1/H2-2/H2-3, H-5/H2-6/H-7/H-20, H-9/H2-11, H-13/H2-14 and H2-21/H3-22 in the 1H-1H COSY spectrum. A linkage of the quaternary C-4 with C-3, C-5, C-18 and C-19 was constructed by two- and three-bond heteronuclear correlation signals from H3-18 and H2-19 to C-3, C-4 and C-5 and from H3-18 to C-19 (Fig. 2) in the HMBC spectrum of 1. A connection of the nitrogen with C-19, C-20, and C-21 was deduced from the HMBC signals of C-20 and C-21 with H2-19, together with their chemical shifts. An attachment of C-1, C-5, C-9 and C-20 to the same quaternary C-10 was determined by the HMBC signals from H-1 to C-9, C-10 and C-20 and from H-9 to C-5. The HMBC signals from H-7 to C-9, from H-9 to C-14, and from H-15 to C-7, C-8 and C-9 revealed a linkage of the quaternary C-8 with C-7, C-9, C-14 and C-15. According to the HMBC signals from H-11b and H-14b to C-12, the quaternary carbon C-12 was inserted between C-11 and C-13, while the HMBC signals from H-17b with C-16 and C-15 revealed that the quaternary C-16 connected by C-15 and C-17. Furthermore, the HMBC signal of C-16 with H-13 and their chemical shifts indicated the presence of an oxygen-bridge between C-13 and C-16. To satisfy requirements of the substitution, chemical shift, and molecular composition, three hydroxy groups were positioned at C-1, C-12 and C-15, respectively, a sulfonic acid group was located at C-17, and one more oxygen-bridge was assigned between C-12 and C-16. Thus, compound 1 was elucidated to have the planar structure as given in Fig. 1, which possesses unique novel 13,16-seco-napelline carbon skeleton and hydroxydioxolane motif formed via the oxygen-bridges between C-16 with C-12 and C-13. The zwitterionic formula was proposed on the basis of the presence of both the acidic and alkali functionalities in the molecule of 1.

|
Download:
|
| Fig. 2. Main 1H-1H COSY (blue bold lines) and HMBC correlations (red arrows, from proton to carbon) of 1 and 2. | |
{kind=link}
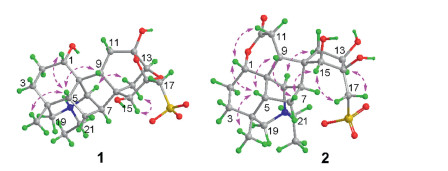
For the relative stereochemistry of 1, a cofacial orientation of H-1, H-5, H-9, and H3-18 on one side of the fused ring system was determined by NOE signals of H-5 with H-1, H-9, H3-18 (Fig. 3) in the NOESY spectrum. The orientation of H2-14, H-15 and H-20 on another side of the fused ring system was deduced from the 2D NOE signals between H-14a and H-20 and between H-14b and H-15. The above spectroscopic deduction, together with restriction of the highly fused ring system, the relative stereochemical structure of 1 was assigned as shown in Fig. 2. Crystallization of 1 in a mixture of MeOH and H2O (3:1) afforded a crystal suitable for crystallographic analysis. Follow-up single crystal X-ray diffraction analysis (anomalous Cu Kα radiation scattering) assigned the absolute configuration of 1 [Flack parameter, 0.006 (12)], an ORTEP diagram shown in Fig. 4. Thus, the structure of compound 1 was unambiguously determined and named aconapelsulfonine A.

|
Download:
|
| Fig. 3. Main 2D NOESY correlations (between the pink double arrowed hydrogens) of 1 and 2. | |
{kind=link}

|
Download:
|
| Fig. 4. ORTEP diagram of 1. | |
{kind=link}
Compound 2, a white amorphous powder, [α]D20 -14.6 (c 0.11, H2O), is an isomer of 1 as indicated by HR-ESIMS and NMR spectroscopic data. When the NMR spectroscopic data of the two compounds were compared (Table 1), replacement of the semi-acetal and acetal carbons in 1 by a lactone carbonyl carbon [δC 178.0 (C-12)] and a hydroxy-bearing quaternary carbon [δC 80.7 (C-16)] in 2 was observed. This observation suggested that 2 had another seco-napelline carbon skeleton to generate the lactone carbonyl carbon, which was verified by 2D NMR spectroscopic data analysis. Besides the signals (Fig. 2) to prove the structural moiety shared by 1 and 2, the 1H-1H COSY correlation signals H-9/H2-11 and the HMBC signals from H-9 and H2-11 to the lactone carbonyl C-12 demonstrated a 12,13-cleavage of the napelline carbon skeleton for 2. Additionally the 1H-1H COSY correlation signals H-13/H2-14 and the HMBC signals from H2-14 to C-7, C-8, C-9, C-15 and C-16; from H-15 to C-7, C-8, and C-9; and from H2-17 to C-13, C-15 and C-16 as well as their chemical shifts proved the presence of a 13,15,16-trihydroxy-17-sulfonate five-membered spiro-ring moiety consisting of C-8 and C-13—C-17 in 2. Although the expected three-bond correlation signal from H-1 to C-12 was unobservable in the HMBC spectrum of 2, the H-1 and C-1 resonances in 2 had larger chemical shift values than that in 1 and related compounds [6j], demonstrating that the lactone ring must be form between the oxygen-bearing C-1 and the carboxylic carbonyl C-12 in 2. The lactone ring formation was supported by the molecular formula as well as a 2D NOE signal between H-1 and H-11a (Fig. 3) in the NOESY spectrum of 2. Moreover, the NOE signals between H-5 with H-1, H-9 and H3-18 and between H-6a and H-9 indicated that these hydrogens spatially oriented on the identical side of the fused rings in 2. The NOE signals between H2-17 with H-13 and H-15 demonstrated that the three hydroxy groups on the spiro-cyclopentane ring had the same orientation. The NOE signals between H-6a and H-15 and between H-14b and H-20 unraveled the relative configuration of the spiro-atom C-8 as shown in Fig. 3. Based on the structural similarity and plausible biogenetic origin (see below) of 2 and 1, the absolute configuration of 2 was assigned to be identical to that of 1, except that the 13S configuration in 1 was reversed to a 13R configuration in 2 due to oxidative cleavage of the C-12—C-13 bond and hydroxylation of C-13. Comparison of the experimentally measured CD and theoretically calculated ECD spectra (Fig. S2 in Supporting information) was supportive to the assignment. Accordingly, the structure of compound 2 was elucidated to possess a 12,13-seco-napelline carbon skeleton with functionalities of 12,1-lactone and 13β, 15β, 16β-trihydroxy-17-sulfonate, and designated as aconapelsulfonine B.
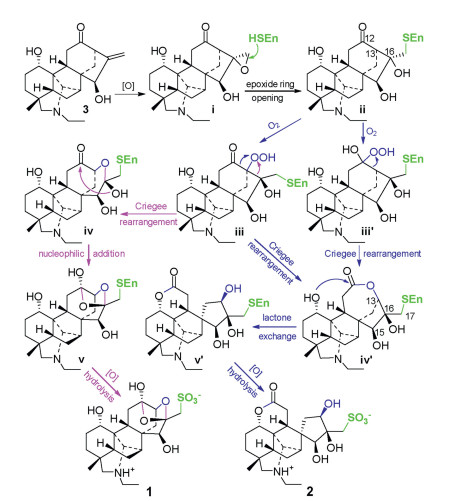
Compounds 1 and 2 are zwitterionic sulfonated C20-diterpenoid alkaloids with different seco-napelline carbon skeletons. From their structural features, songorine (3, Scheme 1), belonging to the napelline-type C20-diterpenoid alkaloid and co-occurring in this plant [6f], is traced to be the most possible biosynthetic precursor. A double bond oxidation of the precursor affords an epoxide ⅰ, then the epoxide ring is enzymatically opened to give an enzyme-adducting product ⅱ. Oxidation of ⅱ at C-13 gives the Criegee intermediate ⅲ, which undergoes either 13,16- and 12,13-bond migrating rearrangements to generate semi-acetal ⅳ and lactone ⅳ′, respectively. An intramolecular nucleophilic addition of ⅳ would afford ⅴ, while a lactone exchange reaction of ⅳ′ would yield ⅴ′. Subsequently the enzyme-adducts ⅴ and ⅴ′ would be broken by oxidative hydrolysis to liberate 1 and 2, respectively. Alternatively, compound 2 may be formed from ⅱ via the Criegee intermediate ⅲ′ of Baeyer-Villige oxidation and successive rearrangement, lactone exchange, and oxidative hydrolysis. In addition, the enzyme-adducts might be broken to liberate the corresponding diterpenoid alkaloid intermediates at any step of the proposed pathways.

|
Download:
|
| Scheme 1. Proposed biogenetic pathways of 1 and 2. | |
{kind=link}
Inspired by clinic application of "Fu Zi" as an analgesic agent [3] and positive responses of our previous work [6g-6j], 1 and 2 were assayed on an acetic acid-induced mice writhing model for the analgesic effect [7] (Supporting information). At dosages of 0.1, 0.3 and 1.0 mg/kg (i.p.), the mice writhing was significantly inhibited in a dose-dependent manner by 58.1%, 63.6% and 70.5% for 1 and 13.8%, 19.3% and 45.5% for 2 [the positive control morphine, 0.3 mg/kg (i.p.), 84.6%].
In conclusion, two analgesic diterpenoid alkaloids with sulfonated zwitterionic properties and undescribed different carbon skeletons were characterized from the water extract of "Fu Zi", supporting the clinic effects and application protocol of the herbal medicine. Biogenetically the skeletal architectures are proposed to be originated from metabolic oxidation accompanied by the Criegee rearrangements of the different carbon bonds in the napelline-type skeleton. The distinctive structures, along with the analgesic effects, are of interesting for future investigations. The proposed biogenetic routes are informative for biomimetic synthesis and structural modification as well as the involving biosynthetic enzymes (Baeyer-Villiger monooxygenases [8]) of the diterpenoid alkaloids.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsFinancial support from the National Natural Sciences Foundation of China (Nos. 81630094 and 81573445), CAMS Innovation Fund for Medical Science (Nos. 2017-I2M-3-010, 2017-I2M-3-011, and 2016-I2M-1-010) and The Drug Innovation Major Project (2018ZX09711001-003-001) is acknowledged.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.09.062.
| [1] |
(a) X.Y. Liu, Y. Qin, Nat. Prod. Rep. 34 (2017) 1044-1050; (b) J.J. Pflueger, L.C. Morrill, J.N. deGruyter, et al., Org. Lett. 19 (2017) 4632-4635; (c) Y. Nishiyama, S. Yokoshina, T. Fukuyama, Org. Lett. 19 (2017) 5833-5835; (d) K.G.M. Kou, J.J. Pflueger, T. Kiho, et al., J. Am. Chem. Soc. 140 (2018) 8105-8109; (e) S. Zhou, R. Guo, P. Yang, et al., J. Am. Chem. Soc. 140 (2018) 9025-9029; (f) N.A. Doering, K.G.M. Kou, K. Norseeda, et al., J. Org. Chem. 83 (2018) 12911-12920; (g) Q.Z. Zhang, Z.S. Zhang, Z. Huang, et al., Angew. Chem. Int. Ed. 57 (2018) 937-941; (h) J. Liu, D. Ma, Angew. Chem. Int. Ed. 57 (2018) 6676-6680; (i) S. Xie, G. Chen, H. Yan, et al., J. Am. Chem. Soc. 141 (2019) 3435-3439; (j) W. Nie, J. Gong, Z. Chen, et al., J. Am. Chem. Soc. 141 (2019) 9712-9718; (k) L. Zou, K. Yu, Y. Fan, et al., ACS Chem. Neurosci. 10 (2019) 1318-1325; (l) S. Shao, H. Xia, M. Hu, et al., J. Neuroinflam. 17 (2020) 13. |
| [2] |
(a) F.P. Wang, Q.H. Chen, X.Y. Liu, Nat. Prod. Rep. 27 (2010) 529-570; (b) Z.Q. Mu, H. Gao, Z.Y. Huang, et al., Org. Lett. 14 (2012) 2758-2761; (c) J.F. Zhang, L. Chen, S. Huang, et al., J. Nat. Prod. 80 (2017) 3136-3142. |
| [3] |
(a) R. Chen, G. Sun, Q. Zhang, et al., China J. Chin. Mater. Med. 38 (2013) 1126-1129; (b) S. Liu, F. Li, Y. Li, et al., J. Ethnopharmacol. 207 (2017) 237-250; (c) J. Wu, Z. Guo, Y. Zhu, et al., Phytomedicine 44 (2018) 187-203; (d) M. Yang, X. Ji, Z. Zou, Toxins 10 (2018) 391. |
| [4] |
(a) L. Wang, J.Y. Ding, X.X. Liu, et al., Acta Pharm. Sin. 49 (2014) 1699-1704; (b) G. Zhou, L. Tang, X. Zhou, et al., J. Ethnopharm. 160 (2015) 173-193; (c) X. Zong, X. Yan, J.L. Wu, et al., J. Nat. Prod. 82 (2019) 980-989; (d) Y.Y. Zhu, G. Yu, Y.Y. Wang, et al., Chem. Nat. Comp. 55 (2019) 189-193; (e) W. Xu, M. Zhang, H. Liu, et al., Nat. Prod. Res. 33 (2019) 1486-1490; (f) T.Q. Do, B.N. Truong, H.D.T. Mai, et al., J. Asian Nat. Prod. Res. 21 (2019) 507-515. |
| [5] |
(a) Q. Guo, C. Xu, M. Chen, et al., Acta Pharm. Sin. B 8 (2018) 933-943; (b) L.J. Meng, Q.L. Guo, C.G. Zhu, et al., Chin. Chem. Lett. 29 (2018) 119-122; (c) L. Meng, Q. Guo, M. Chen, et al., Chin. Chem. Lett. 29 (2018) 1257-1260; (d) Q. Guo, D. Li, C. Xu, et al., Acta Pharm. Sin. B 10 (2020) 895-902. |
| [6] |
(a) B. Jiang, S. Lin, C. Zhu, et al., J. Nat. Prod. 75 (2012) 1145-1159 1878, and 2008; (b) Z.B. Jiang, B.Y. Jiang, C.G. Zhu, et al., J. Asian Nat. Prod. Res. 16 (2014) 891-900; (c) Z.B. Jiang, X.H. Meng, B.Y. Jiang, et al., Chin. Chem. Lett. 26 (2015) 653-656; (d) X.H. Meng, Z.B. Jiang, C.G. Zhu, et al., Chin. Chem. Lett. 27 (2016) 993-1003; (e) X.H. Meng, Z.B. Jiang, Q.L. Guo, et al., Chin. Chem. Lett. 28 (2017) 588-592; (f) X.H. Meng, Q.L. Guo, C.G. Zhu, et al., Chin. Chem. Lett. 28 (2017) 1705-1710; (g) Q. Guo, H. Xia, G. Shi, et al., Org. Lett. 20 (2018) 816-819; (h) Q. Guo, H. Xia, X. Meng, et al., Acta Pharm. Sin. B 8 (2018) 409-419; (i) Y. Wu, S. Shao, Q. Guo, et al., Org. Lett. 21 (2019) 6850-6854; (j) Q. Guo, H. Xia, Y. Wu, et al., Acta Pharm. Sin. B 10 (2020) 1954-1965. |
| [7] |
Q. Chen, Experimental Methodology of Pharmacolog. Beijing: People's Health Press, 2010.
|
| [8] |
(a) I. Polyak, M.T. Reetz, W. Thiel, J. Am. Chem. Soc. 134 (2012) 2732-2741; (b) B.J. Yachnin, T. Sprule, M.B. McEvoy, et al., J. Am. Chem. Soc. 134 (2012) 7788-7795; (c) G. Li, M. Garcia-Borras, M.J.L.J. Furst, et al., J. Am. Chem. Soc. 140 (2018) 10464-10472. |