 2021, Vol. 32
2021, Vol. 32 


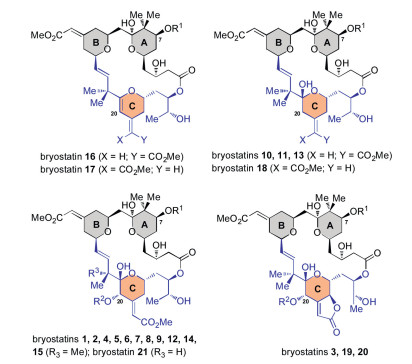
The bryostatins are well-known marine natural products produced by a bacterial symbiont of the bryozoan Bugula neritina (Scheme 1) [1]. Since Pettit and co-workers discovered bryostatin 1 in 1982 [2], these molecules have captured wide attention of the chemical, biological and medical communities because of their promising potential in the treatment of numerous diseases and conditions such as cancer [3], diabetes [4], ischemic stroke [5], Alzheimer's disease [6] and HIV [7]. The bryostatins family consists of 21 congeners. While their northern part (A and B rings) differ from each other only in the C7-acyl residues, the C ring nucleus in the southern part displays significant diversity including both the overall oxidation states and the C20-acyl substitution (Scheme 1). It has been suggested that the unique biological activity of the bryostatins is associated with the solvent-exposed portion of the A and B rings, while the C ring simply binds to the C1 domain of isoforms of protein kinase C [8]. Despite of this assertion, several impressive C ring analogs have been developed by simplifying the C17, C20 and C26-positions [9], indicating the C ring could be a potential target motif for developing superior byrostatin analogs.

|
Download:
|
| Scheme 1. Structures of bryostatins 1-21. | |
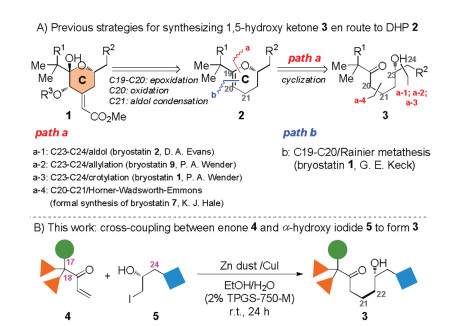
In the accomplished syntheses of bryostatins [10], five groups utilized dihydropyran (DHP) 2 as a general precursor [10a-c, i, k], which was transformed into the C ring nucleus 1 via sequential operations of epoxidation/oxidation/aldol condensation condensation (Scheme 2A). DHP 2 was typically accessed by acid-catalyzed cyclization of 1,5-hydroxyl ketones 3, except Keck used an intramolecular Rainier metathesis reaction between ester and vinyl groups to form the C19-C20 double bond [10a]. To synthesize 3, Evans used aldol reaction in the synthesis of bryostatin 2 [10c], and Wender used asymmetric allylation and crotylation in the syntheses of bryostatins 9 [10k] and 1 [10b], respectively, to install the stereogenic C23-hydroxyl by forming the C23-C24 bond from the corresponding 1,5-keto aldehyde. In contrast, Hale utilized Horner-Wadsworth-Emmons reaction to form the C20-C21 bond of 1,5-hydroxyl ketone 3 in the formal synthesis bryostatin 7 [10i].

|
Download:
|
| Scheme 2. Previous strategies to form the C-ring (A); Cross-coupling between α-hydroxy iodide and enone to form 1,5-hydroxyl ketones (B). | |
In our continuing studies of bryostatins [10j], we have been seeking a general method, which not only enables a convergent synthesis of the C ring itself, but also offering an efficient access to diverse C ring analogs. Herein, we report a convergent entry into the C ring precursor 1,5-hydroxy ketone 3 (Scheme 2B). The approach relies on a Zn/Cu-promoted conjugate addition [11] of α-hydroxy iodides 5 with enones 4 by modifying the protocols invented by Lipshutz [12] and Luche [13] independently. The free hydroxyl group in 5 was found to improve the coupling in aqueous media [14] remarkably, while previous work typically used protected hydroxy halides. The reaction proceeds under mild conditions with no need for preforming air- or moisture-sensitive organometallic precursors, leading to direct formation of the C21-C22 bond with good tolerance of diverse functionalities at the C17-, C18- and C24-positions (numbering according to bryostatins). The synthetic value of the approach was also demonstrated by a four-step transformation of 3 into the known intermediate in our total synthesis of bryostatin 8.
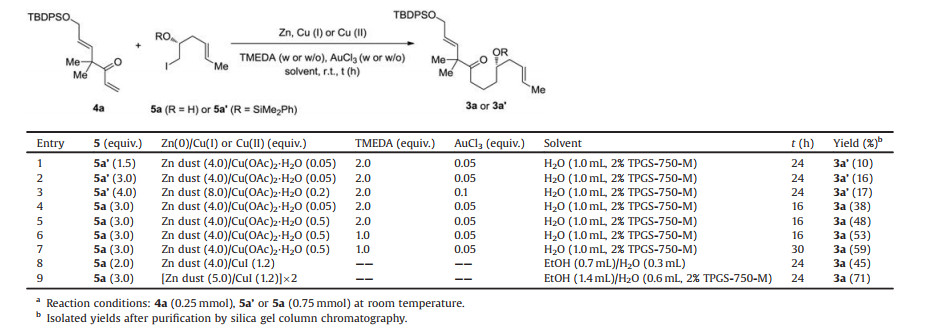
Enone 4a was initially tested in the conjugation addition with α-silyloxy iodide 5a'. 4a was synthesized from the commercially available 2,2-dimethyl-pent-4-enoic acid methyl ester by four steps in an overall yield of 66% [10a]. 5a was synthesized from commercially available (R)-(-)-benzyl glycidyl ether by five steps in an overall yield of 77% [15]. 5a was silylated to give 5a' in 96% yield.
We first employed Lipshutz' micellar protocol, which was developed in water with 2% TPGS-750-M as nonionic surfactant. In the presence of 4.0 equiv. of Zn dust and 0.05 equiv. of Cu(OAc)2·H2O along with 2.0 equiv. of TMEDA and 0.05 equiv. of AuCl3 as additives, the desired adduct 3a' was obtained, but only in 10% yield with recovery of a large amount of 4a (Table 1, entry 1). Increasing the loading of 5a' from 1.5 equiv. to 3.0 equiv. slightly improved the yield to 16% (entry 2). We also tested increasing the loading of 5a', Zn, Cu(OAc)2·H2O and AuCl3 together (entry 3). Unfortunately, the operation did not improve the efficiency at all. To our surprise, the unprotected α-hydroxy iodide 5a showed a much higher reactivity than 5a', leading to 3a in 38% yield (entry 4). Probably, the hydrophilic hydroxyl group favors 5a to form the nanomicelles in water within which the addition occurs, while the hydrophobic silyloxy group in 5a' disfavors such function. Another possible reason might be that the bulky silyloxy group sterically inhibited the addition. Using 5a as the substrate, the yield of 3a was further improved to 48% by increasing the loading of Cu(OAc)2·H2O from 0.05 equiv. to 0.5 equiv. (entry 5). We also found that less amount of TMEDA (entry 6) and longer reaction time (entry 7) provided a higher yield of 59%. To further simplify the reaction conditions, Luche's protocol using 4.0 equiv. of Zn and 1.2 equiv. of CuI without any additives was examined. 3a was obtained in 45% yield in a mixed solvent of EtOH/H2O (7:3) (entry 8). We finally tested the efficiency of the protocol developed by combining both Lipshutz's and Luche's methods. Thus, the reaction was performed in EtOH/H2O containing 2% TPGS-750-M with adding extra portion of Zn (5.0 equiv.)/CuI (1.2 equiv.) after 12 h. This operation gave 3a in an optimal yield of 71% (entry 9).
|
|
Table 1 Screening the cross-coupling conditions.a |
{kind=link}
{kind=link}
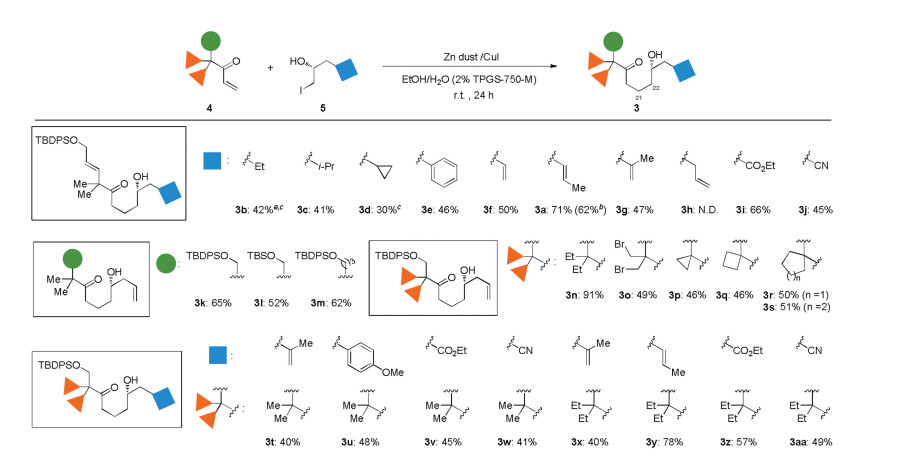
The scope of the C24-functionality in α-hydroxy iodide 5 was first examined with enone 4a under the optimal reaction conditions (Scheme 3). The approach was suitable for synthesizing branched and cyclic, or aryl substitution (3b-3e), except 3d containing a cyclopropane was obtained in 30% yield. The 98:2 er of 3b and 95:5 er of 3d indicated that the coupling conditions did not interfere with the chirality of the secondary hydroxy moiety. The alkenyl substitution, which could be transformed into the C25-C26 dihydroxyl moiety in bryostatins, was also tolerated to give 3a, 3f and 3g. In contrast, the allylic substitution disfavored the coupling, affording a complex mixture (3h). The low efficiency might be owing to the competitive 5-exo-trig cyclization [16] of the initially formed α-hydroxy radical with the allyl group. The success of forming of 3i and 3j showcased the promising value of enriching the diversity of the C-ring analogs, because the ester and nitrile groups could be further transformed into a variety of functionalities. Besides the allyloxy substitution, the C17-functionality was expanded to silyl protected hydroxymethyl or hydroxypropyl group, providing 3k-3m in 52%-65% yields. The approach also showcased the power of creating the impressive diversity at the C18-positon, which has been barely investigated. Compared with dimethyl-substituted 3l, diethyl-substituted 3n was delivered in a higher yield of 91%, suggesting that the steric hinderance at the C18-positon in enone might favor the coupling. The reaction showed a chemoselectivity for coupling with iodomethyl group, as the geminal bromomethyl group was survived to give 3o in 49% yield. Enones 4 with C18-cyclic substituents ranging from small rings to media rings served as good substrates to give 3p-3s. Various combinations of C18- and C24-functionality were finally examined, providing 3t-3aa in mediate to good yields.

|
Download:
|
| Scheme 3. Scope of cross-coupling. Reaction conditions: 4 (0.25 mmol), 5 (0.75 mmol), [Zn (5.0 equiv.)/CuI (1.2 equiv.)]×2,0 h and 12 h one for each, EtOH (1.4 mL), H2O (2% TPGS-750-M) (0.6 mL), room temperature, 24 h. a Isolated yields after purification by silica gel column chromatography. b The yield in parentheses was obtained on 500 mg-scale of 4a. c The 98:2 er of 3b and 95:5 er of 3d were determined by HPLC analysis using a chiral stationary phase. | |
{kind=link}
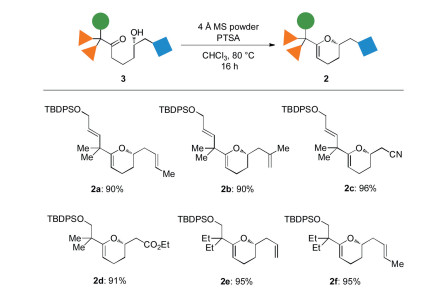
The resulting 1,5-hydroxy ketones 3 underwent an acid-catalyzed cyclization to give the corresponding DHP 2a-2f in high yields. As shown by the representative examples in Scheme 4, the functionality at the C17, C18 and C24-postions did not affect the good cyclization efficiency.

|
Download:
|
| Scheme 4. Cyclization of 3 to DHP 2. Reaction conditions: 3 (0.2 mmol), PTSA (0.002 mmol) and 4Å MS (100 mg) in 2 mL of CHCl3 at 80 ℃ for 16 h. Isolated yields after purification by silica gel column chromatography. | |
{kind=link}
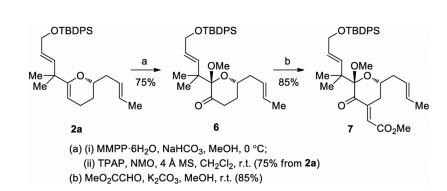
The approach was further applied in the concise synthesis of 7, the known intermediate in our total synthesis of bryostatin 8 (Scheme 5). DHP 2a underwent epoxidation of the C19-C20 double bond with MMPP·6H2O in MeOH, followed by in situ epoxide ring opening at the C19 position to give the ketal alcohol. Without purification, the crude ketal alcohol was oxidized with TPAP/NMO to deliver pyranone 6 in an overall yield of 75% from 2a. Aldol condensation of 6 with methyl glyoxylate introduced the exo-cyclic E-enoate, giving 7 in 85% yield.

|
Download:
|
| Scheme 5. Synthesis of 7. | |
{kind=link}
In summary, we have developed a convergent approach to 1,5-hydroxy ketones, the general C ring precursor for synthesizing bryostatins. The approach proceeds via a Zn/Cu-promoted conjugate addition of α-hydroxy iodides with enones, leading to 1,5-hydroxy ketones by direct formation of C21-C22 bond with good tolerance of diverse functionalities at the C17-, C18- and C24-positions. The resulting 1,5-hydroxy ketone was transformed into the known intermediate in our total synthesis of bryostatin 8. Application of the approach in the synthesis of the C ring analogs and the downstream medical application are undergoing in our group.
Declaration of competing interestThe authors report no declarations of interest.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (No. 21921002), the National Science and Technology Major Project of the Ministry of Science and Technology of the People's Republic of China (No. 2018ZX09711001-005-004), and the Fundamental Research Funds for the Central Universities (No. 2012017yjsy210). The authors thank Mr. Wenqin Yang for his invaluable help in this work.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.11.039.
| [1] |
(a) K.J. Hale, S. Manaviazar, Chem. Asian J. 5 (2010) 704-754; (b) X.R. Tian, H.F. Tang, X.L. Tian, et al., Future Med. Chem. 10 (2018) 1497-1514; (c) R. Wu, H. Chen, N. Chang, et al., Chem. Eur. J. 26 (2020) 1166-1195; (d) R. Raghuvanshi, S.B. Bharate, Curr. Top. Med. Chem. 20 (2020) 1124-1135. |
| [2] |
G.R. Pettit, C.L. Herald, D.L. Doubek, et al., J. Am. Chem. Soc. 104 (1982) 6846-6848. DOI:10.1021/ja00388a092 |
| [3] |
(a) M.R. Farlow, R.E. Thompson, L.J. Wei, et al., J. Alzheimers Dis. 67 (2019) 555-570; (b) J. Wang, Z. Wang, Y. Sun, et al., Biochem. Biophys. Res. Commun. 512 (2019) 473-478. |
| [4] |
K.J. Way, N. Katai, G.L. King, Diabet. Med. 18 (2001) 945-949. DOI:10.1046/j.0742-3071.2001.00638.x |
| [5] |
K. Mizutani, S. Sonoda, H. Wakita, et al., Neuroreport. 27 (2016) 659-664. DOI:10.1097/WNR.0000000000000592 |
| [6] |
(a) T.J. Nelson, M.K. Sun, C. Lim, et al., J. Alzheimer's Dis. 58 (2017) 521-535; (b) T. Sarajärvi, M. Jäntti, K.M.A. Paldanius, et al., Neuropharmacology 141 (2018) 76-88; (c) K. Murakami, M. Yoshimura, S. Nakagawa, et al., Int. J. Mol. Sci. 21 (2020) 1179-1191. |
| [7] |
(a) M.D. Marsden, B.A. Loy, X.M. Wu, et al., PLoS Pathog. 13 (2017)e1006575; (b) M.Z. Zhao, E.D. Crignis, C. Rokx, et al., Pharmacol. Res. 139 (2019) 524-534; (c) E.F. Heffern, R. Ramani, G. Marshall, et al., J. Virus Erad. 5 (2019) 84-91; (d) A. Proust, C. Barat, M. Leboeuf, et al., J. Glia 68 (2020) 2212-2227; (e) B.X. Li, H. Zhang, Y. Liu, et al., Sci. Rep. 10 (2020) 3511-3522. |
| [8] |
J.M. Ketcham, I. Volchkov, T.Y. Chen, et al., J. Am. Chem. Soc. 138 (2016) 13415-13423. DOI:10.1021/jacs.6b08695 |
| [9] |
(a) B.M. Trost, H. Yang, O.R. Thiel, et al., J. Am. Chem. Soc. 129 (2007) 2206-2207; (b) N. Kedei, M.B. Kraft, G.E. Keck, et al., J. Nat. Prod. 78 (2015) 896-900; (c) D. Staveness, R. Abdelnabi, K.E. Near, et al., J. Nat. Prod. 79 (2016) 680-684; (d) D.O. Baumann, K.M. McGowan, N. Kedei, et al., J. Org. Chem. 81 (2016) 7862-7883; (e) P.R. Mears, S. Hoekman, C.E. Rye, et al., Org. Biomol. Chem. 17 (2019) 1487-1505; (f) M.B. Kraft, Y.B. Poudel, N. Kedei, et al., J. Am. Chem. Soc. 136 (2014) 13202-13208; (g) I.P. Andrews, J.M. Ketcham, P.M. Blumberg, et al., J. Am. Chem. Soc. 136 (2014) 13209-13216; (h) J.L. Sloane, N.L. Benner, K.N. Keenan, et al., Proc. Natl. Acad. Sci. U.S. A. 117 (2020) 10688-10698; (i) C. Hardman, S. Ho, A. Shimizu, Nat. Commun. 11 (2020) 1879-1889. |
| [10] |
(a) G.E. Keck, Y.B. Poudel, T.J. Cummins, et al., J. Am. Chem. Soc. 133 (2011) 744-747; (b) P.A. Wender, C.T. Hardman, S. Ho, et al., Science 358 (2017) 218-223; (c) D.A. Evans, P.H. Carter, E.M. Carreira, et al., Angew. Chem. Int. Ed. 37 (1998) 2354-2359; (d) D.A. Evans, P.H. Carter, E.M. Carreira, et al., J. Am. Chem. Soc. 121 (1999) 7540-7552; (e) K. Ohmori, Y. Ogawa, T. Obitsu, et al., Angew. Chem. Int. Ed. 39 (2000) 2290-2294; (f) B.M. Trost, Y.L. Wang, A.K. Buckl, et al., Science 368 (2020) 1007-1011; (g) M. Kageyama, T. Tamura, M.H. Nantz, et al., J. Am. Chem. Soc. 112 (1990) 7407-7408; (h) Y. Lu, S.K. Woo, M.J. Krische, J. Am. Chem. Soc. 133 (2011) 13876-13879; (i) S. Manaviazar, M. Frigerio, G.S. Bhatia, et al., Org. Lett. 8 (2006) 4477-4480; (j) Y.B. Zhang, Q.Y. Guo, X.W. Sun, et al., Angew. Chem. Int. Ed. 57 (2018) 942-946; (k) P.A. Wender, A.J. Schrier, J. Am. Chem. Soc. 133 (2011) 9228-9231; (l) B.M. Trost, G. Dong, Nature 456 (2008) 485-488; (m) B.M. Trost, G. Dong, J. Am. Chem. Soc. 132 (2010) 16403-16416. |
| [11] |
(a) P. Knochel, R.D. Singer, Chem. Rev. 93 (1993) 2117-2188; (b) A. Alexakis, J.E. Bäckvall, N. Krause, et al., Chem. Rev. 108 (2008) 2796-2823; (c) F. Zhou, X. Hu, W. Zhang, et al., Org. Chem. Front. 5 (2018) 3579-3584; (d) J.A. Shin, J. Kim, H. Lee, et al., J. Org. Chem. 84 (2019) 4558-4565; (e) Y. Ouyang, Y. Peng, W.D.Z. Li, Tetrahedron 75 (2019) 4486-4496. |
| [12] |
(a) A. Krasovskiy, C. Duplais, B.H. Lipshutz, Org. Lett. 12 (2010) 4742-4744; (b) C. Duplais, A. Krasovskiy, B.H. Lipshutz, Organometallics 30 (2011) 6090-6097; (c) B.H. Lipshutz, S. Ghorai, W.W.Y. Leong, et al., J. Org. Chem. 76 (2011) 5061-5073; (d) B.H. Lipshutz, S.L. Huang, W.W.Y. Leong, et al., J. Am. Chem. Soc. 134 (2012) 19985-19988; (e) H. Pang, Y. Wang, F. Gallou, et al., J. Am. Chem. Soc. 141 (2019) 17117-17124. |
| [13] |
(a) C. Petrier, C. Dupuy, J.L. Luche, Tetrahedron Lett. 27 (1986) 3149-3152; (b) J.L. Luche, C. Allavena, Tetrahedron Lett. 29 (1988) 5369-5372; (c) J.L. Luche, C. Allavena, C. Petrier, Tetrahedron Lett. 29 (1988) 5373-5374; (d) C. Dupuy, C. Petrier, L.A. Sarandeses, et al., Synth. Commun. 21 (1991) 643-651; (e) L.A. Sarandeses, A. Mourinoband, J.L. Luche, J. Chem, Soc. Chem. Commun. 11 (1992) 798-799. |
| [14] |
(a) C.J. Li, Chem. Rev. 105 (2005) 3095-3165; (b) C.J. Li, L. Chen, Chem. Soc. Rev. 35 (2006) 68-82; (c) Y. Kim, C.J. Li, Green. Synth. Catal. 1 (2020) 1-11. |
| [15] |
J.S. Yadav, M.S. Reddy, A.R. Prasad, Tetrahedron Lett. 46 (2005) 2133-2136. DOI:10.1016/j.tetlet.2005.01.121 |
| [16] |
J.E. Baldwin, R.C. Thomas, L.I. Kruse, et al., J. Org. Chem. 42 (1977) 3846-3852. DOI:10.1021/jo00444a011 |