 2020, Vol. 31
2020, Vol. 31 


b Traditional Chinese Medicine Department, The First Affiliated Hospital, Sun Yat-sen University, Guangzhou 510080, China;
c Department of Nephrology, Shenzhen Traditional Chinese Medicine Hospital, Guangzhou University of Chinese Medicine, Shenzhen 518033, China;
d School of Pharmaceutical Sciences, Guangdong Provincial Key Laboratory of New Drug Screening, Southern Medical University, Guangzhou 510515, China
Hepatocellular carcinoma (HCC) is the most common form of primary liver cancer and second leading cause of cancer-associated death worldwide [1]. The clinical management of advanced and metastatic HCC is challenging in many countries. Unfortunately, at diagnosis less than 30% of patients are deemed eligible for curative interventions including surgical resection, liver transplantation, and chemoembolization. Moreover, conventional chemotherapeutic drugs such as doxorubicin have not been shown to significantly extend the survival of the patients with HCC [2, 3]. Other conventional chemotherapeutic drugs such as cisplatin, 5-fluorouracil, or their combinations therein were found to have even lower efficacy than doxorubicin alone [4-6]. Many ongoing clinical trials aim to improve the treatment of HCC by targeting specific subpopulations of patients. In 2007, sorafenib, was approved by the FDA as a first-line treatment for advanced HCC, but the treatment only increased progression-free survival by a paltry two months compared to placebo [7]. Meanwhile, over 80% of phase three trials for targeted molecular therapies with in the last five years have failed to prolong survival of patients with advanced diseases. Therefore, it is of great clinical significance to develop novel and efficient therapeutic strategies against HCC due to the lack of target selectivity, conventional systemic chemotherapies.
Triptolide (TP), a diterpenoid triepoxide isolated from the plant Tripterygium wilfordii, is a natural product that has already been found to be highly eff; ective against many malignant cancer types including pancreatic cancer, liver cancer, and cholangiocarcinoma [8-10]. TP causes liver cancer cell death in vitro and in vivo by the induction of apoptosis. Therefore, TP is a potential therapeutic agent. Although TP was initially considered to be a promising chemotherapeutic agent, its potential clinical application has been limited due to its poor solubility and extremely high toxicity.
Nanotechnology-based drug delivery systems have been shown to improve tumor drug therapeutically efficiency and reduce toxicity effect [11-17]. Nano-medicines are emerging as the next-generation anticancer agents [18-26]. Nanoparticles can be designed to be responsive to the local tumor microenvironment or to tissue-specific inter actions with selectively over-expressed receptors in tumors [27-29]. This can include "active targeting" of tumor cells surface receptors (e.g., folate receptor) [30] or drug release triggered by the intracellular enzymes. Moreover, the extracellular pH of solid tumors is more acidic (pH ~6.8) in comparison to normal tissue, and the endolysosomes of cancer cells are even more acidic (pH < 6) [31].
Moreover, it is reported that redox balance in the tumor cells is higher with more reduced reductive agents such as glutathione (GSH) and reactive oxygen species such as H2O2 [32, 33]. Utilization of the different GSH level in tumor can be considered as a robust and simple strategy to achieve tumor microenviromently responsive drug release [34]. Herein, we report on the design and synthesis of a tumor-responsive nanoformulated triptolide (Nf-TP) for HCC targeted therapy (Scheme 1).

|
Download:
|
| Scheme 1. Schematic illustration of preparation, cell uptake, synchronous intracellular drug release, and mechanism of action. | |
In this case, we prepare a new polymer drug carrier for TP delivery. Firstly, we polymerized the isocyanate and bis(2-hydroxyethyl)-disulfide, and then the hydrophilic PEG as the end block was added to prepare a new amphiphilic polymer (PSSP) which contained both hydrophobic parts and hydrophilic heads. This polymer was further systematically characterized by 1H NMR (Fig. S1 in Supporting information). The result demonstrated the successful synthesis of the polymer carrier.
We then dissolved 10 mg PSSP and 2 mg TP in anhydrous DMF (800 μL). After the mixture was stirring for 30 min, the deionized water (2.4 mL) was added dropwise in it and to prepare the nanoparticles (PSSP@TP). The nanoparticles were further purified by dialysis (MW = 3500 Da).
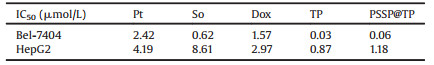
According to the TEM images in Fig. 1A, the shape of PSSP@TP is perfect spherical. According to our previous measurement on DLS, the size was 186±10 nm (PDI < 0.3). Then PSSP@TP was treated with 10 mmol/L GSH for 7 h. The TEM and DLS images (Figs. 1B and C) revealed the complete disassembly of NPs. These results confirmed that the PSSP@TP would disassemble and the TP would release in the reductive environment. Furthermore, we monitored the stability of PSSP@TP in H2O and 10% FBS at 37 ℃ for 7 days. Base on the results, we did not detect significant change in size or PDI, proving that the nano-system was stable enough (Fig. S2 in Supporting information). Drug-loading capacity (DL) and encapsulation efficiency (EE) are the key factors in drug delivery. To obtain high drug loading and reach the therapeutic needs, we processed the optimized formulations to prepare for PSSP@TP. As mentioned above, the PSSP@TP has a satisfactory DL of 2%±0.76%; whilst EE is only 16%±5.6%. The cleavage of polymer chain by GSH can cause the break of PSSP@TP to release TP (Scheme S1 in Supporting information). Then we set up two experimental groups to conduct drug release experiments: PSSP@TP and PSSP@TP with 10 mmol/L GSH, to continuously monitor the release of PSSP@TP. Fig. 1D showed that PSSP@TP released TP rapidly in the presence of 10 mmol/L GSH. The existence of GSH can significantly accelerate the release of PSSP@TP.

|
Download:
|
| Fig. 1. Characterizations of PSSP@TP. Typical TEM images of PSSP@TP (A) and PSSP@TP with 10 mmol/L GSH (B) for 7 h; (C) DLS intensity of PSSP@TP before and after treated with 10 mmol/L GSH for 7 h; (D) Time and GSH dependent TP release profiles of PSSP@TP in deionized water at GSH 0 and 10 mmol/L, pH 7.4. Scale bars: 200 nm. | |
To determine the mechanism and characterize the intracellular uptake of NPs, we started to load the PSSP with the hydrophobic fluorescent dye rhodamine B (RhB) and incubated Bel-7404 cells with PSSP@RhB for 4 h and 7 h. Confocal laser scanning microscopy (CLSM) were used to observe cells (Fig. 2A). According to the image from CLSM, the red fluorescence signal intensity increased over time from 4 h to 7 h in Bel-7404 cells. Subsequently, we used flow cytometry to monitor and quantify the uptake process (Fig. 2B). The uptake of Bel-7404 cells showed a 99-fold sharply increase from 0 h to 7 h, indicating effective intracellular uptake (Fig. 2C). To investigate the cytotoxicity of PSSP@TP in vitro, MTT assays were performed to verify the cell viability in human live cancer lines Bel-7404 and HepG2. These cells were treated with five different drugs for 48 h: cisplatin (Pt), sorafenib (So), doxorubicin (Dox), TP and PSSP@TP. The IC50 values are shown in Table 1. We found that PSSP@TP showed higher cytotoxicity than Pt, So and Dox. The IC50 value of Bel-7404 cells treated with PSSP@TP for 48 h showed a 40-fold decrease as compared with Pt. In addition, the IC50 value of HepG2 incubated with PSSP@TP exhibited an 8-fold decrease in contrast to So. In vitro evaluation of anticancer activity of nanoparticles was performed as shown in Fig. 3. In general, the killing effect of PSSP@TP on hepatoma cell lines Bel-7404 and HepG2 was significantly higher than that of Pt, So and Dox (Figs. 3A and B).

|
Download:
|
| Fig. 2. In vitro evaluation of intracellular uptake of nanoparticles by fluorescence. (A) CLSM of 7404 cells after incubation with PSSP@RhB for 4 h, 7 h. The cell nucleus was stained blue by Hoechst 33342. The actin cytoskeleton was stained green by Phalloidin FITC. The PSSP@RhB fluorescence was marked as red. Scale bar: 20 μm. Flow cytometry (B) and statistics (C) of cellular uptake of NPs after treated with PSSP@RhB for 1 h, 4 h and 7 h. | |
|
|
Table 1 IC50 values of Pt, So, Dox, TP and PSSP@TP on different cell lines. |
{kind=link}
{kind=link}
{kind=link}

|
Download:
|
| Fig. 3. In vitro evaluation of anticancer activity of nanoparticles. Relative cell viability of 7404 (A) and HepG2 (B) cells after 48 h incubation of Cisplatin, Sorafenib, Doxorubicin, TP, PSSP@TP. Cell apoptosis images of 7404 (C) and HepG2 (D) cells treated with PBS, Cisplatin, Sorafenib, Doxorubicin, TP, PSSP@TP for 24 h. Image J was performed to determine the total percentages of 7404 (E) and HepG2 (F) cells that underwent apoptosis. Data are represented as mean±standard deviation (SD; n = 3, t-test; ***P < 0.001). | |
{kind=link}
With regard to the toxicity of TP to normal cells, we have done the following experiments to verify this problem. LO2 is a normal hepatocyte, which can be represented by normal cells. The toxicity of TP and PSSP@TP to it was detected. The results of MTT experiments showed that the toxicity of PSSP@TP to normal cells was much less than that of TP, Pt and So (Fig. S3 in Supporting information).
With the intention of further study on the apoptosis effect of drugs, we utilized flow cytometry to analyse cell apoptosis. In the Bel-7404 cell line, after 48 h of treatment, the percentage of apoptosis in PBS, Pt, So, Dox, TP and PSSP@TP groups were 2.5%, 4.096%, 13%, 7.582%, 45.15%, 45.93%, respectively. These results indicated that TP and PSSP@TP increased the apoptosis rate of the Bel-7404 cells. It follows that PSSP@TP has a strong effect on promoting apoptosis and necrosis of Bel-7404 cells. The same conclusion can be reached in HepG2 cell lines (Figs. 3C and D). Together, the relative apoptosis rate of the five groups, the relative apoptosis rate of PSSP@TP group is much higher than that of PBS, Pt, So and Dox groups (Figs. 3E and F).
Next, PSSP@Cy7.5 was prepared and employed to characterize the biodistribution of PSSP@TP. BALB/c nude mice bearing patientderived xenografts (PDX) of liver cancer (PDLC) were imaged at 1, 10 and 24 h after tail vein administration of PSSP@Cy7.5. More PSSP@Cy7.5 accumulated in the tumor site over time (Fig. 4A). According to the tumor and major organs fluorescence in vitro, the fluorescence intensity of the tumor was stronger than that of other organs except the liver (Figs. 4B and C), which was due to the EPR effect of nanoparticles.

|
Download:
|
| Fig. 4. Tumor imaging and biodistribution of PSSP@Cy7.5 nanoparticles in vivo. (A) Tumor-bearing nude mice injected with PSSP@Cy7.5 nanoparticles through tail vein at different time points before IVIS imaging. (B) The ex vivo images. (C) Fluorescence intensity of major organs collected from mice at 24 h post injection of PSSP@Cy7.5 nanoparticles. The data are shown as mean±SD (n = 3). | |
{kind=link}
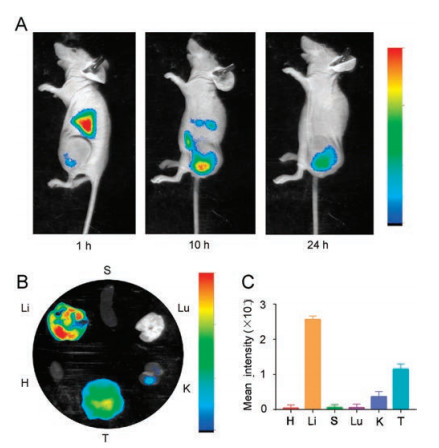
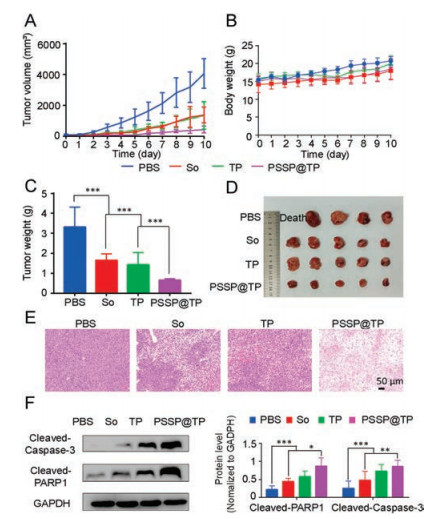
Subsequently, a PDLC model was established to evaluate the anticancer efficacy of PSSP@TP. Compared with the other drugs, PSSP@TP displayed the highest antitumor efficacy among all groups (Fig. 5A). The mice in PSSP@TP-treated group did not show significant loss in body weight (Fig. 5B). After 11 days, we observed that the average tumor volume of the PBS group increased to over 3000 mm3, while the tumor growth was well confined in the PSSP@TP group (431 mm3). In the end, all mice were sacrificed. Their tumors and normal tissues were collected and weighed. The mice treated with PSSP@TP had the smallest tumors (Fig. 5C). It was only 1/10 of the PBS treated group and 1/3 of TP and So groups (Fig. 5D).

|
Download:
|
| Fig. 5. In vivo antitumor efficacies. (A) Patient-derived cancer xenograft (PDX) models of liver tumor growth curves. (B) Body weight changes of mice. (C) Tumor weight of mice. (D) Photos of partial PDX models of liver tumors. (E) H&E analyses of tumor tissues collected from the mice administrated with various formulations. Scale bar is 50 μm. (F) Cellular total proteins were extracted. Apoptotic proteins cleaved caspase-3 and cleaved PARP1 were detected by western blot analysis. The data are expressed as means±SD (n = 3, t-test; *P < 0.05, **P < 0.01, ***P < 0.001). GADPH is the abbreviation of glyceraldehyde-3-phosphate dehydrogenase. | |
{kind=link}
Pathological analysis further confirmed the antitumor effect of PSSP@TP. The liver tumors of nude mice treated with PSSP@TP showed extensive nuclear atrophy and disappearance by H&E staining (Fig. 5E), proving that PSSP@TP had good anticancer effects. H&E sections of other organs showed no significant tissue changes at the effective dose (Fig. S4 in Supporting information), which may be due to the short treatment time. After that, the fresh tissues of each group were used for western blot assay.
The whole tissue lysate was extracted for western blot assay. Cleaved caspase-3 and cleaved PARP1 in PSSP@TP group were significantly up-regulated (Fig. 5F), confirming that triptolide induced caspase-3-dependent apoptosis and that the PSSP@TP was more toxic to tumor tissue.
Eleven days after tail vein injection, retro-orbital blood collection was performed to determine the central liver and kidney function index of nude mice. The heart, liver and kidney function indexes of PSSP@TP were closer to that of PBS-treated group (Fig. S5 in Supporting information). However, the free drug treated mice showed significant changes in CK-MB and ASTof heart and liver function indexes. These results indicated that nanoparticles group did not cause changes in heart, liver and kidney function, but could significantly inhibit the tumor growth.
In conclusion, we create an evolutional polymer composite which could not only polymerize the isocyanate and bis(2-hydroxyethyl)-disulfide, but also more importantly create a reductive responsiveness to GSH in the tumor microenvironment. Results showed that the TP could rapidly release from nanoparticles in reductive conditions. PSSP@TP induced higher toxicity and increased cell apoptosis in vitro. Compared with only single TP composite, PSSP@TP improved drug antitumor efficacy and lower systemic toxicity in vivo. Furthermore, PSSP@TP induced higher levels of cell apoptosis as compared to TP. In summary, we reported a novel strategy for triptolide delivery based on microenvironment responsiveness polymer composite, which provided a potential method for cancer therapy.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 81572797, 81701817), the Natural Science Foundation of Guangdong Province (No.2019A1515011619), Guangdong Provincial Science and Technology Department (No. 2016A030311015), and Shenzhen Science and Technology Project (No. JCYJ20180507183842516).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.05.034.
| [1] |
D. Dimitroulis, C. Damaskos, S. Valsami, et al., World J. Gastroenterol. 23 (2017) 5282-5294. DOI:10.3748/wjg.v23.i29.5282 |
| [2] |
M.A. Avila, C. Berasain, B. Sangro, et al., Oncogene 25 (2006) 3866-3884. DOI:10.1038/sj.onc.1209550 |
| [3] |
Y. Sun, W. Ma, Y. Yang, et al., Asian J. Pharm. Sci. 14 (2019) 581-594. DOI:10.1016/j.ajps.2019.04.005 |
| [4] |
Y. Cheng, P. Zhao, S. Wu, et al., Int. J. Pharm. 545 (2018) 261-273. DOI:10.1016/j.ijpharm.2018.05.007 |
| [5] |
Y. Ding, S. Li, W. Ge, et al., Eur. J. Med. Chem. 183 (2019) 111706.
|
| [6] |
T. Kawaoka, H. Aikata, T. Kobayashi, et al., Hepatol. Res. 48 (2018) 1118-1130. DOI:10.1111/hepr.13232 |
| [7] |
J.M. Llovet, S. Ricci, V. Mazzaferro, et al., N. Engl. J. Med. 359 (2008) 378-390. DOI:10.1056/NEJMoa0708857 |
| [8] |
P.A. Phillips, V. Dudeja, J.A. McCarroll, et al., Cancer Res. 67 (2007) 9407-9416. DOI:10.1158/0008-5472.CAN-07-1077 |
| [9] |
D. Ling, H. Xia, W. Park, et al., ACS Nano 8 (2014) 8027-8039. DOI:10.1021/nn502074x |
| [10] |
T. Tengchaisri, R. Chawengkirttikul, N. Rachaphaew, et al., Cancer Lett. 133 (1998) 169-175. DOI:10.1016/S0304-3835(98)00222-5 |
| [11] |
W. Ma, S.N. Sha, P.L. Chen, et al., Adv. Healthc. Mater. 9 (2019) e1901100.
|
| [12] |
Y. Yu, Q. Xu, S. He, et al., Coord. Chem. Rev. 387 (2019) 154-179. DOI:10.1016/j.ccr.2019.01.020 |
| [13] |
Y. Xiao, F. An, J. Chen, et al., Small 15 (2019) e1903121. DOI:10.1002/smll.201903121 |
| [14] |
Y. Cao, Z. Wei, M. Li, et al., Curr. Cancer. Drug. Targets 19 (2019) 338-347. DOI:10.2174/1568009618666180629150927 |
| [15] |
Q. Hu, L. Bai, Z. Zhu, et al., Chin. Chem. Lett. 31 (2020) 915-918. DOI:10.1016/j.cclet.2020.01.008 |
| [16] |
Z. Chen, C. Wu, Z.F. Zhang, et al., Chin. Chem. Lett. 29 (2018) 1601-1608. DOI:10.1016/j.cclet.2018.08.007 |
| [17] |
Y. Sun, A. Zhan, S. Zhou, et al., Chin. Chem. Lett. 30 (2019) 1435-1439. DOI:10.1016/j.cclet.2019.05.001 |
| [18] |
X. Pu, L. Zhao, J. Li, et al., Acta Biomater. 88 (2019) 357-369. DOI:10.1016/j.actbio.2019.02.039 |
| [19] |
Y. Yang, X. Wang, G. Liao, et al., J. Colloid Interf. Sci. 509 (2017) 515-521. |
| [20] |
L. Zong, X. Li, H. Wang, et al., Int. J. Pharm. 531 (2017) 108-117. DOI:10.1016/j.ijpharm.2017.08.082 |
| [21] |
H. Jin, C. Wan, Z. Zou, et al., ACS Nano 12 (2018) 3295-3310. DOI:10.1021/acsnano.7b08148 |
| [22] |
H. Wang, Z. Lu, L. Wang, et al., Cancer Res. 24 (2017) 6963-6974. |
| [23] |
W. Zhang, X. Hu, Q. Shen, et al., Nat. Commun. 10 (2019) 1704. DOI:10.1038/s41467-019-09566-3 |
| [24] |
C. Yang, L. Xing, X. Chang, et al., Mol. Pharm. 17 (2020) 1300-1309. DOI:10.1021/acs.molpharmaceut.9b01318 |
| [25] |
Y. Yang, Q. Chen, J. Lin, et al., Curr. Med. Chem. 26 (2019) 2285-2296. DOI:10.2174/0929867326666190409130207 |
| [26] |
X. Pu, L. Zhao, J. Li, et al., Acta Biomater. 88 (2019) 357-369. DOI:10.1016/j.actbio.2019.02.039 |
| [27] |
M. Yu, D. Su, Y. Yang, et al., ACS Appl. Mater. Inter. 11 (2019) 176-186. DOI:10.1021/acsami.8b16219 |
| [28] |
Y. Liu, Y. Kim, N. Siriwon, et al., Biotechnol. Bioeng. 115 (2018) 1403-1415. DOI:10.1002/bit.26566 |
| [29] |
Y. Yang, X. Wang, G. Liao, et al., J. Colloid Interf. Sci. 509 (2017) 515-521. |
| [30] |
Z. Soe, B. Poudel, T. Nguyen, et al., Asian J. Pharm. Sci. 14 (2019) 40-51. DOI:10.1016/j.ajps.2018.09.004 |
| [31] |
Y. Li, W. Hong, H. Zhang, et al., J. Control. Release 317 (2020) 232-245. DOI:10.1016/j.jconrel.2019.11.031 |
| [32] |
Q. Chen, Y. Yang, X. Lin, et al., Chem. Commun. (Camb.) 54 (2018) 5369-5372. DOI:10.1039/C8CC02791A |
| [33] |
W. Tao, Z. He, Asian J. Pharm. Sci. 13 (2018) 101-112. DOI:10.1016/j.ajps.2017.11.002 |
| [34] |
X. Xu, P. Saw, W. Tao, et al., Adv. Mater. 29 (2017) 1700141. DOI:10.1002/adma.201700141 |