 2020, Vol. 31
2020, Vol. 31 


b School of Bioengineering, Dalian University of Technology, Dalian 116024, China;
c Department of Neurosurgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai 200127, China
The sprouting discoveries of genomic tools (e.g., plasmid DNA, small interference RNA, microRNA, clustered regularly interspaced short palindromic repeats genome editing systems: CRISPR) herald the upcoming era of gene therapy as versatile therapeutics for treatment of a multitude of intractable diseases [1-3]. Nonetheless, the bottleneck that impedes stride of gene therapy into wide clinical availabilities is acknowledged to lack well-defined gene delivery systems as viral vectors for transportation of nucleic acids cargos to appropriate intracellular compartments [4, 5]. It should be noted that, despite the appealing transcellular scenarios contrived by viral vectors, their intrinsic drawbacks including potential immunogenicity, limited nucleic acids loading capacities, difficulties in scale-up [6], encouraged researchers to elaborate well-defined synthetics to circumvent a string of biological barriers and accomplish gene transportation mission. In principle, exogenous gene delivery systems, once being administered into physiological community, series of eliminating machineries are postulated to orchestrate collectively to implement clearance of the foreign species either by reticuloendothelial (RES) system triggered by protein adsorption (and opsonization) or degradation of genomic payloads through enzymatic degradation by nucleases (naked DNA was reported to be susceptible to thorough digestion in bloodstream within seconds) [7-10]. Aiming to resolve these issues, the therapeutic nucleic acids are imperative to accommodate inside the protective carriers with excellent stealthiness as possible, preventing accessibility of the nucleases and other biological species that potentially commit to interactions with or destabilization onto the architecture of delivery systems (resulting in exposure of encapsulated nucleic acids to nucleases). To acquire this stealthiness, the surface chemistry of delivery systems is apparently critical to modulate. To this respect, surface modification by poly(ethylene glycol) (PEGylation) onto biomedical devices, for instance: nanoscaled delivery systems in particular, is deemed to be a superior strategy for minimizing adverse non-specific biointerfacial reactions, accounting for dispersion of PEGylated nanoparticles in the bloodstream to stealthily circulate [11, 12]. Nonetheless, the external PEG shell has also been verified to elicit reluctant affinities to the cytomembrane as well [13, 14]. Therefore, limited cellular uptake activities were extensively reported for PEGylated gene/drug delivery systems, which hold responsibilities for the ultimate inadequate pharmaceutic efficacy despite substantial accumulation in pathological sites accredited to the aforementioned PEGylation strategy. Learning from this PEGylation dilemma, it is conceivable that detachment of PEGylation (dePEGylation) in subsequence to accumulation at the pathological sites should be crucial in stimulating subsequent transcellular delivery and accomplishing the ultimate pharmaceutic consequence. Aim for selective detachment of PEGylation at pathological sites, PEGylation through a labile linkage responsive to the pathologic microenvironment could represent a rational approach. Of note, recent molecular biology revealed the crucial role of matrix metalloproteinases (MMPs) in the process of tumor invasion and metastasis [15]. Particularly, MMP-2 has been elucidated to be uniformly overexpressed among spectra of tumors, which is responsible to degrade extracellular type Ⅳ collagen by following sequence-specific manners [16]. Hence, in the present study, we proposed a peptide linkage (whose amino acids sequence specifically assigned to MMP-2) between the blocks of PEG-block-poly{N'-[N-(2-aminoethyl)-2-aminoethyl]aspartamide} [PAsp(DET)], wherein the polycationic PAsp(DET) segments could self-assemble with negative-charged nucleic acids into nanoscaled core based on electrostatic interactions. Yet, PEG segments tethered onto PAsp(DET) through MMP-2-degradable peptide linkage represent as the external biocompatible shells. Moreover, folic acids (FA) whose receptors are overexpressed on the surface of tumor cells were installed at the distal end of PEG with the aim of targeted delivery into tumors [17]. Therefore, in subsequence of folic acids-mediated tumor accumulation, it could be envisioned that the overexpressed MMP-2 in tumors could readily cleave the proposed peptide linkage, thus committing dePEGylation, and facilitating the subsequent cellular endocytosis of the remaining unPEGylated polycations-based nanoparticles. It should also be noted that the proposed PAsp(DET) has appreciable endosome escape capacity due to its facilitated protonation function in acidic endosomes. Therefore, our proposed system could represent an intriguing strategy to circumvent the dilemma of PEGylation, and shed important implications in promoting transcellular/subcellular delivery of therapeutic cargos for tumor therapy.
The peptide-linked block copolymer of FA-PEG-GPLGVRG-PAsp (DET) was synthesized according to synthetic scheme in Fig. 1a. In brief, heterobifunctional linear PEG (N3-PEG-FA) was schemed to conjugate with alkynyl-functionalized peptide (alkynyl-GPLGVRG) through classic click chemistry reaction. The successful peptide conjugation was confirmed by FT-IR measurement, as evidenced by complete disappearance of characteristic absorbance from azido group at approximate 2104 cm–1 (Fig. S1 in Supporting information). Furthermore, the distal amino group of peptide was employed as the initiator for polymerization of monomer of NCABLA to yield FA-PEG-GPLGVRG-PBLA. Eventually, cationic DET components were attached onto the synthesized PBLA segment through aminolysis for production of the ultimate FA-PEG-GPLGVRG-PAsp(DET). The synthesized block copolymer of FA-PEG-GPLGVRG-PAsp(DET), as well as the precursors of FA-PEG-GPLGVRG and FA-PEG-GPLGVRG-PBLA were characterized by 1H NMR measurement (Fig. 1b, Figs. S2 and S3 in Supporting information) and the polymerization of PAsp(DET) segments were determined to be approximate 69. In addition, control polymers of PEG-PAsp(DET) (void of both labile peptide linkage and targeting ligand of FA) and PEG-GPLGVRG-PAsp(DET) (void of targeting ligand of FA) were also synthesized by following similar synthetic procedures. The yielded polymers were characterized by 1HNMR measurement and the detailed information in pertinent to their chemical structures was summarized in Table 1.

|
Download:
|
| Fig. 1. Illustration of synthetic route of FA-PEG-GPLGVRG-PAsp(DET) (a) and its 1H NMR spectrum (b). | |
|
|
Table 1 Chemical descriptions of a class of block copolymers. |
A class of polyplex micelles were prepared by electrostatic complexation between the synthesized cationic block copolymer and anionic pDNA at varied N/P ratios (defined as the molar ratio of positive charges from the amino groups of the block copolymer and the negative charges from phosphate groups of pDNA). The electrostatic complexation behaviors were investigated by gel electrophoresis. Overall, all the synthesized block copolymers, including PEG-PAsp(DET), PEG-GPLGVRG-PAsp(DET) and FA-PEG-GPLGVRG-PAsp(DET), could effectively neutralize the negative charges of pDNA (Fig. S4 in Supporting information). There seems no distinctive difference in their charge-neutralizing capacities, and complete neutralization appeared to start at N/P ratio of 1. In consistent with gel electrophoresis results, zeta potential measurements also confirmed fully neutralization when N/P ratios exceed 1, wherein relative positive surface net charge (ranging from +3.5 mV to +5.5 mV) for the prepared polyplex micelles (Table 2, no significant difference was determined for the polyplex micelles at varied N/P ratios), in contrast to naked pDNA being approximate –31.2 mV in zeta potential, approved the effective electrostatic neutralization behaviors. It should be also note that homopolymer of PAsp(DET)-based polyplex exhibited markedly higher surface net charge, approximate +29.6 mV in zeta potential, again verifying the functional impact of PEGylation on charge-masking of electrostatic self-assemblies, thereby speculated to afford reduced reactions with charged biological species or biological structures in physiological environment. Furthermore, the self-assembled formation was transferred for DLS measurement and the corresponding results were summarized in Table 2. Overall, approximate 100 nm or sub-100 nm nanostructures were determined with unimodal size distribution. Also, there was no significant difference in particle size between the polyplex micelles prepared from PEG-PAsp(DET), PEG-GPLGVRG-PAsp (DET) and FA-PEG-GPLGVRG-PAsp(DET), yet relative smaller size was attainable at higher N/P ratios. This is not surprising to consider electrostatic complexation between pDNA and block copolymer being thermodynamic process. Higher N/P ratios representing a larger number of block polymers surrounding pDNA, thereby allows for fast charge-neutralization process. Consequently, the rapid gain in entropy due to charge-neutralization could be converted as higher degree condensing potency to pDNA. Aside from DLS measurement, the pDNA condensation morphologies by PEG-GPLGVRG-PAsp(DET) was characterized by TEM measurement. As shown in Fig. 2a, distinctive worm-like nanoparticles were identified rather than the conventional spherical nanoparticles. Indeed, this observation is consistent with our previous results for PEG-PLys-based polyplex micelle [18]. Note that DNA is a rather rigid macromolecule, whose persistence length is determined to be approximate 50 nm in the physiological condition [19]. Therefore, DNA condensation could adopt a regular folding scheme rather than complete collapse-down into spherical structures.
|
|
Table 2 Physiochemical characterizations of diverse polyplex micelles. |
{kind=link}

|
Download:
|
| Fig. 2. Physiochemical characterizations of self-assemblies of PEG-GPLGVRG-PAsp (DET) and pDNA at N/P ratio of 8: Morphologies of PEG-GPLGVRG-PAsp(DET)-based polyplex micelle (a) in absence and (c) in presence of MMP-2 (characterized to be spherical nanoparticles with obvious aggregation). Scale bar: 100 nm; (b) Time-dependent measurement for PEG-GPLGVRG-PAsp(DET)-based polyplex micelle in presence or absence of MMP-2. | |
{kind=link}
To explore the feasibilities of dePEGylation function, polyplex micelle composed of PEG-GPLGVRG-PAsp(DET) was incubated in presence of MMP-2 [MMP(+)], and the reaction solution was transferred for real-time DLS measurement. As shown in Fig. 2b, as opposed to the constant DLS for polyplex micelle from PEG-GPLGVRG-PAsp(DET) in absence of MMP-2 [MMP(-)], progressive growth in DLS size was observed for [MMP(+)] starting from the initial 100 nm in DLS diameter to become almost micrometer scale. To dissect the underlying of this structural transition, TEM measurement was conducted for [MMP(+)]. As shown in Fig. 2c, apparent secondary aggregation between nanoparticles was constantly observed for [MMP(+)]. A plausible reason for this aggregation should be as a consequence of MMP-2-mediated peptide cleavage, which induced the departure of PEG shell from the electrostatically interacted PAsp(DET) and pDNA compartments. Lack of this spatial PEG exterior, the inter-nanoparticle association between electrostatic interaction-based self-assemblies would become facilitated. Therefore, the rising occurrence of inter-particle aggregates could be anticipated to account for the observed rapid growth in DLS measurement. In addition to identification of secondary aggregations, another noteworthy is the observation of relatively spherical DNA condensates post dePEGylation in contrast to the aforementioned worm-like DNA condensates. Our previous studies delineated the crucial role of the tethered PEG chains in mediating pDNA packaging [20], wherein electrostatically interactions for compartmentation of polycations and pDNA will inevitably result in unfavorable crowding of the tethered PEG chains as the external corona. Apparently, this PEG crowding hinders segmental motion of PEG chains, which is unfavorable with respect to the restricted conformation motion of PEG. Therefore, the tethered PEG segments would represent as counteract factor to prevent DNA from collapse-down, vice versa, dePEGylation should behave as an additional drive force to encourage higher-degree pDNA condensation into the observed spherical structures. To this end, the obtained results approved readily dePEGylation function upon enzymatic degradation from our elaborated polyplex micelle, and the biological impact of this dePEGylation function was investigated hereafter.
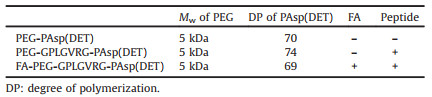
As suggested in DLS measurement, dePEGylated DNA delivery nanoconstructs could elicit non-specific association for selfaggregation (Fig. 2b). To this respect, it is conceivable that dePEGylated nanoconstructs should also confer improved nonspecific affinities to the cell membranes, thereby facilitating a larger number of pDNA payloads internalized inside the cells. To verify this speculation, the cellular uptake efficiency of polyplex micelle from PEG-peptide-PAsp(DET) w/o MMP-2 treatment was evaluated in HeLa cells. As shown in Fig. 3, reluctant cellular uptake was confirmed for polyplex micelle without MMP-2 treatment, affirming the drawbacks of PEGylated nanoparticles [PEG-GPLGVRG-PAsp(DET)] in rendering inferior affinities to cell membranes. The ligand of FA appeared to promote cellular uptake efficiency (Figs. 3a and b), which is believed as a result of the FA receptors overexpressed on the surface of cancerous cells (e.g., Hela). Note that the promotion seems to be particularly obvious for Hela cells in FA(-) medium (Fig. 3a), suggesting the validity of FAmediated promotion in cellular uptake for FA-PEG-GPLGVRG-PAsp(DET)-based polyplex micelle. To our interest, the cellular uptake efficiency appeared to experience pronounced jump for polyplex micelle with MMP-2 treatment (approximate 3–4 fold, Figs. 3c and d), which should be accredited to improved affinities due to dePEGylation, exceeding the promotive index by FA ligand.

|
Download:
|
| Fig. 3. Impact of dePEGylation on cellular uptake efficiencies. (a) Cellular uptake of polyplex micelles in DMEM without FA. Grey (PBS), green [polyplex micelle from PEG-GPLGVRG-PAsp(DET)], purple [polyplex micelle from FA-PEG-GPLGVRG-PAsp (DET)] and orange [polyplex micelle from FA-PEG-GPLGVRG-PAsp(DET) post treatment of MMP-2]. (b) Cellular uptake of polyplex micelles in DMEM with FA. (c) Cellular uptake efficiency of PEG-GPLGVRG-PAsp(DET)-based polyplex micelle w/o MMP-2 treatment in HeLa cells; (d) Representative CLSM images of PEG-GPLGVRG-PAsp(DET)-based polyplex micelle at N/P ratio of 8 w/o MMP-2 treatment. Scale bar: 10 μm. | |
{kind=link}
The nanoscaled particles were documented to internalize inside the cells through endocytotic pathway, where the internalized substances would entrap in the acidic endosomes and further digestive lysosomes. Therefore, gene delivery systems have to endow adequate facilities to retrieve the vulnerable payloads from lysosome entrapment. Pertaining to endosome escape, our proposed dePEGylation strategy, as well as pH-responsive protonation function of PAsp(DET) is essential. As reported previously cationic materials are conjectured to escape from endosome entrapment by means of their direct destabilization on the structures of endosome membranes. Particularly, the destabilizing potencies were verified to follow a distinctive charge density-dependent manner [21], namely, higher positive-charge densities could impose more powerful disrupting potency to the negatively charged cytomembranes.
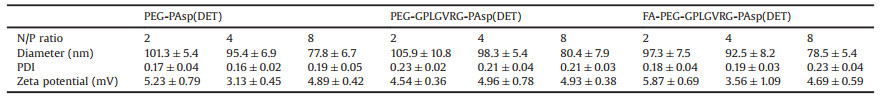
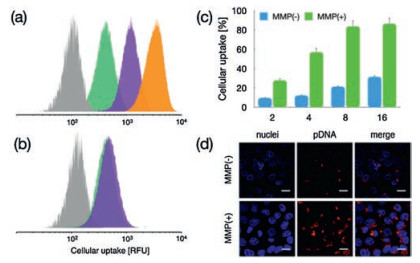
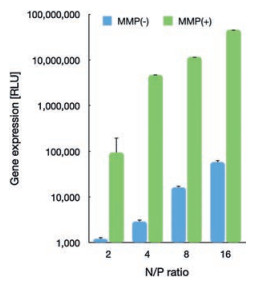
To this respect, the promoted protonation of amino groups of PAsp(DET) in acidic endosome (pH 5.5, protonation percentage: approximate 80%) [21], relative to its in neutral physiologic environment (pH 7.4, protonation percentage: 50%), could transform into highly charged polycations to exert endosome disruption. Due to the charge-masking effect from PEGylation, the direct interactions between PAsp(DET) and endosome membranes should be minimal. Yet, our elaborated dePEGylation strategy could resolve this unfavorable spatial barrier of PEGylation. Hence, amplified association and destabilization onto endosome membranes could be concluded. To verify this hypothesis, the membrane destabilizing potency was assessed for PEG-GPLGVRG-PAsp(DET) w/o MMP-2 treatment at pH 5.5 based LDH assay. In light of LDH as intracellular enzyme, quantification of the extracellular LDH for the cells upon sample treatment could represent as a valid indicator for estimation of the loss of membrane integrity. As shown in Fig. 4c, minimal extracellular LDH was detected for MMP(-), suggesting its limited interactive potency to endosome membrane and excellent biocompatibility when PEGylation remained. On the contrary, spike in extracellular levels of LDH were confirmed for MMP(+), indicating the functional impact of dePEGylation on intensifying the destabilizing potencies to cytoplasm membranes. Relying on this powerful potency, the entrapment of MMP(+) in endosomes/lysosomes were determined to be remarkably lower (Fig. 4a), as evidenced by significantly lower colocalization ratio of MMP(+) with endosomes/lysosomes (approximate 0.23) relative to the colocalization ratio of MMP-2(-) (approximate 0.65) (Fig. 4b). The facilitated cellular uptake and promoted endosome escape ultimate contributed to markedly high gene expression activities at the affected cells. As shown in Fig. 5, the gene expression level of MMP-2(+) is approximate one- or two-order magnitude higher than MMP-2(-). To this end, the obtained result demonstrated the success of our dePEGylation strategy in seeking promoted gene delivery at the targeted cells.

|
Download:
|
| Fig. 4. (a) Confocal laser scanning microscopy (CLSM) observation for intracellular trafficking of PEG-GPLGVRG-PAsp(DET)-based polyplex micelle in HeLa cells: upper image: MMP(-), lower image: MMP(+), red: pDNA, green: endosomes and lysosomes, blue: nuclei. Scale bar: 100 μm. (b) Endosome entrapment was determined by quantification of colocalization ratio of pDNA and endosome/lysosome as summarized in bar graph (**P < 0.01); (c) LDH for assessment of membrane destabilization of PEG-GPLGVRG-PAs colocalization ratio of pDNA and endosome/lysosome as summarized in bar graph (**P < 0.01); (c) LDH for assessment of membrane destabilization of PEG-GPLGVRG-PAs | |
{kind=link}

|
Download:
|
| Fig. 5. Gene expression of PEG-GPLGVRG-PAsp(DET)-based polyplex micelles (containing RFP reporter gene) w/o MMP-2 treatment in HeLa cells. | |
{kind=link}
Aside from promoted gene expression, it is also important to ascertain the safety profile of our constructed system. As shown in Fig. S5 (Supporting information), negligible cytotoxicities were confirmed for all the polyplex micelles from a class of block copolymers. In addition, hemolysis assay also confirmed excellent biocompatibilities, as evidenced by their minimal hemolytic activities Fig. S6 (Supporting information). Moreover, the cytomembrane disrupting potency of MMP(+), despite of dePEGylation, remained limited at physiological pH (Fig. S7 in Supporting information). This appreciable safety profile should be accredited to the pH-dependent protonation function of PAsp(DET), whose protonation at physiological pH is significant lower than endosome pH. The lower charge density of PAsp(DET) should present reduced destabilizing potency to the cytomembranes. This pH-dependent cytomembrane disrupting behaviors is considered to be informative for further design of safe and efficient gene delivery materials.
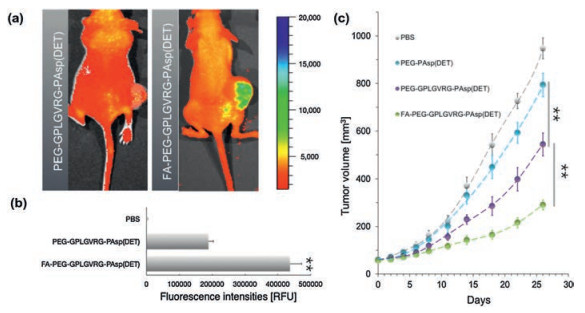
Eventually, the constructed polyplex micelle from FA-PEG-GPLGVRG-PAsp(DET) was attempted for systemic treatment of glioma. Note that glioma remains one of the highest fatalities among diverse spectrums of intractable tumors. Aiming for antiglioma therapy, an anti-angiogenic strategy by means of inhibiting neo-vasculature formation was employed in this study for pursuit of inhibitory growth of glioma. Herein, anti-vascular endothelial growth factor (VEGF) pDNA-encoding soluble fms-like tyrosine kinase 1 (sFlt-1), specifically for inhibiting the signal transduction of VEGF, was used to construct gene delivery system. Therefore, target silencing of VEGF in the tumors can inhibit the angiogenetic activity, consequently capable of suppressing the supply of nutrient or oxygen for tumor growth. Herein, the developed polyplex micelle from PEG-GPLGVRG-PAsp(DET) and FA-PEG-GPLGVRG-PAsp(DET) were intravenously into the bloodstream of xenografted glioma-bearing Bal b/c nude mice via tail vein. As shown in Fig. 6a, markedly higher tumor accumulation was determined for FA-PEG-GPLGVRG-PAsp(DET)-based polyplex micelle relative to PEG-GPLGVRG-PAsp(DET) (approximate 2.4 fold, Fig. 6b) at 12 h post intravenous administration, approving our strategic use of FA as active tumor-docking ligand for promoted tumor-targeting function. In consequence, significant more potent tumor growth suppression was achieved by FA-PEG-GPLGVRG-PAsp(DET)-based polyplex micelle (Fig. 6c). It is also worthy to note that higher inhibitory effect was also identified for PEG-GPLGVRG-PAsp(DET)-based polyplex micelles as compared to PEG-PAsp (DET), which again validates the proposed dePEGylation due to selective GPLGVRG hydrolysis in tumors to seek enhanced gene expression at the targeted tumors and leadingly accomplish potent gene therapy. To this end, the obtained results verified the systemic efficacy of our proposed gene delivery system, which shed important implications for broad utilities in tumor gene therapy.

|
Download:
|
| Fig. 6. In vivo performance of the proposed FA-PEG-GPLGVRG-PAsp(DET)-based polyplex micelle containing anti-angiogenic sFlt-1 pDNA for systemic treatment of xenografted glioma. (a) Polyplex micelles containing Cy5-labeled pDNA for insight onto the biodistribution at 12 h post intravenous dosage by IVIS; (b) The quantified fluorescence intensities at tumors (**P < 0.01); (c) Anti-tumor efficacy by means of measurement of tumor volume (**P < 0.01). | |
{kind=link}
Stimuli-responsive materials are particularly important in design of gene/drug delivery systems in light of distinctive microenvironment during the journey of gene transportation [22-26]. In the present study, precise detachment of PEGylation in responsive to tumor-overexpressed enzymes was demonstrated in our well-defined synthetic gene delivery nanoconstructs. Particularly, dePEGylation has showed sequential benefits in transcellular and subcellular trafficking of nucleic acids payloads. These structural transformations responsive to microenvironment in facilitating gene delivery appeal to the reminiscence of the natural virus, e.g. adenovirus, capable of executing structural transformation to mitigate the encountered biological barriers [27]. Considering the rising knowledge of molecular biology, our proposed enzymatically responsive strategy could provide an even more precise approach, rather than the conventional stimuli (e.g., pH, redox or temperature [28]), to mimicking intelligent virus for efficient gene transfection, which might ignite the advance of nonviral synthetic gene delivery systems as potent therapeutics toward broad clinical availability.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis research was funded by the National Natural Science Foundation of China (No. 21878041). Q. Chen acknowledges the funding support from Talent Project of Revitalizing Liaoning (No. XLYC1807184) and Dalian Science & Technology Innovation Fund (Nos. 2020JJ26SN050, 2020JJ26GX025).
Appendix A. Supplementary datSupplementary material related to this article canbefound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.07.027.
| [1] |
L. Cong, F.A. Ran, D. Cox, et al., Nature 339 (2013) 819-823. |
| [2] |
G.J. Hannon, Nature 418 (2002) 244-251. DOI:10.1038/418244a |
| [3] |
U. Lachelt, E. Wagner, Chem. Rev. 115 (2015) 11043-11078. DOI:10.1021/cr5006793 |
| [4] |
Y. Lee, K. Kataoka, Adv. Polym. Sci. 249 (2012) 95-134. DOI:10.1007/12_2011_129 |
| [5] |
X. Guo, L. Huang, Acc. Chem. Res. 45 (2012) 971-979. DOI:10.1021/ar200151m |
| [6] |
F. Mingozzi, K.A. High, Nat. Rev. Genet. 12 (2011) 341-355. DOI:10.1038/nrg3027 |
| [7] |
K. Miyata, N. Nishiyama, K. Kataoka, Chem. Soc. Rev. 41 (2012) 2562-2574. DOI:10.1039/C1CS15258K |
| [8] |
D.W. Pack, A.S. Hoffman, S. Pun, et al., Nat. Rev. Drug Discov. 4 (2005) 581-593. DOI:10.1038/nrg3027 |
| [9] |
S.K. Samal, M. Dash, S. Van Vlierberghe, et al., Chem. Soc. Rev. 41 (2012) 7147-7194. DOI:10.1039/c2cs35094g |
| [10] |
Y.N. Yue, C. Wu, Biomater. Sci. 1 (2013) 152-170. DOI:10.1039/C2BM00030J |
| [11] |
J.A. Hubbell, A. Chilkoti, Science 337 (2012) 303-305. DOI:10.1126/science.1219657 |
| [12] |
J. Kopecek, Adv. Drug Deliv. Rev. 65 (2013) 49-59. |
| [13] |
Q. Chen, K. Osada, T. Ishii, et al., Biomaterial 33 (2012) 4722-4730. DOI:10.1016/j.biomaterials.2012.03.017 |
| [14] |
S. Takeda, K. Miyata, M. Oba, et al., J. Am. Chem. Soc. 130 (2008) 6001-6009. DOI:10.1021/ja800336v |
| [15] |
N. Johansson, M. Ahonen, V.M. Kähäri, Cell. Mol. Life Sci. 57 (2000) 5-15. DOI:10.1007/s000180050495 |
| [16] |
J. Xiong, H. Gao, J. Microencapsul. 34 (2017) 440-453. |
| [17] |
Y. Lu, P.S. Low, Adv. Drug Deliv. Rev. 64 (2012) 342-352. DOI:10.1016/j.addr.2012.09.020 |
| [18] |
K. Osada, H. Oshima, D. Kobayashi, et al., J. Am. Chem. Soc. 132 (2010) 12343-12348. DOI:10.1021/ja102739b |
| [19] |
G.S. Manning, Biophys. J. 91 (2006) 3607-3616. DOI:10.1529/biophysj.106.089029 |
| [20] |
T.A. Tockary, K. Osada, Q. Chen, et al., Macromolecules 46 (2013) 6585-6592. DOI:10.1021/ma401093z |
| [21] |
K. Miyata, M. Oba, M. Nakanishi, et al., J. Am. Chem. Soc. 130 (2008) 16287-16294. DOI:10.1021/ja804561g |
| [22] |
C. Chen, Y. Li, X. Yu, et al., Chin. Chem. Lett. 29 (2018) 1609-1612. |
| [23] |
J. Peng, Q. Yang, Y. Xiao, et al., Adv. Funct. Mater. 29 (2019) 1900004. |
| [24] |
J. Peng, T. Qi, J. Liao, et al., Nanoscale 4 (2012) 2694-2704. DOI:10.1039/c2nr30147d |
| [25] |
Q. Zhu, M. Saeed, R. Song, et al., Chin. Chem. Lett. 31 (2020) 1051-1059. |
| [26] |
Y. Song, D. Li, J. He, et al., Chin. Chem. Lett. 30 (2019) 2027-2031. |
| [27] |
M.D. Weitzman, R.M. Linden, Adeno-associated virus biology, in: R. Snyder, P. Moullier (Eds.), Adeno-Associated Virus. Methods in Molecular Biology (Methods and Protocols), Vol 807, Humana Press, Totowa, 2012, pp. 1-23.
|
| [28] |
M. Karimi, A. Ghasemi, P.S. Zangabad, et al., Chem. Soc. Rev. 45 (2016) 1457-1501. |