 2020, Vol. 31
2020, Vol. 31 


Cancer is a critical illness which seriously threatens the health and quality of human life [1]. Traditional cancer treatments including radiotherapy and chemotherapy demonstrate significant toxicity side effects. Especially, patients will build up drug resistance after several courses of chemotherapy [2, 3]. Consequently, new approaches against cancer via photodynamic therapy (PDT) [4, 5], photothermal therapy (PTT) [6, 7] and chemodynamic therapy (CDT) [8, 9] attract increasingly attentions. Among them, PDT and PTT require light irradiation, which hinders their applications for cancer treatment in deep tissue due to limited penetration of light [10]. Differently, CDT, firstly defined by Bu and coworkers, kills cancer cells via producing hydroxyl radicals (·OH) by chemical reactions under the stimulation of tumor microenvironment (TME) without the external energy [11]. Specific TME in tumors rather than in normal tissues, including overexpressed hydrogen peroxide (H2O2) and glutathione (GSH) as well as mildly acidic conditions, provide specific conditions for CDT. The Fenton reaction, producing ·OH from H2O2 catalyzed by transition metal ions, has been widely used for CDT. For instance, Lin and coworkers prepared a MnO2 coated mesoporous silica nanoparticles (MS@MnO2 NPs), which effectively generated ·OH and consumed GSH by reacting with MnO2 [12]. Zheng et al. synthesized a coreshell structure network (MON)-p53 via the coordination between Fe3+ and tannic acid [13]. In the system, p53 inhibited the expression of the SLC7A11 (a cystine/glutamate antiporter), resulting in GSH depletion to further enhance the ·OH generation. However, the H2O2 content is not enough for effective CDT although its content in cancer cells is higher than that in normal cells [14]. Thus, to enhance the generation of H2O2 in cancer cells is of great importance for achieving efficient CDT.
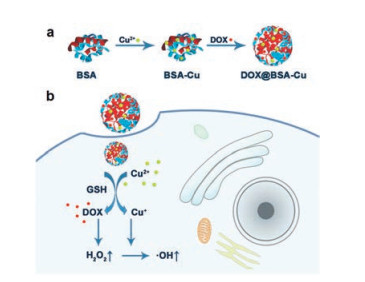
On the other hand, anthracycline antineoplastic antibiotics have been widely used in clinic and research, such as doxorubicin (DOX) [15, 16]. DOX can intercalate into DNA base pairs and influence the replication of DNA, resulting in obvious curative effect on cancer cells [17]. However, DOX also causes serious side effects to healthy tissues when bring toxicity to cancer cells during chemotherapy. DOX at low concentrations that is not toxic to cells can induce the production of reactive oxygen species (ROS), such as H2O2. The moiety of anthracene in the chemical structure of DOX can form semiquinone which would reduce O2 to O2·-, and finally result in the generation of H2O2 via reductase-mediated reactions in cells [18, 19]. Therefore, we proposed that DOX with a low concentration may be used as a supplement for H2O2 in CDT to enhance the treatment efficiency. It is noteworthy that low concentration of DOX used for H2O2 generation can also avoid toxic side effects, thus improve the biosafety for cancer treatment. Herein, we designed DOX@BSA-Cu NPs (nanoparticles) containing bovine serum albumin (BSA), Cu(II) and low-dose DOX, in which Cu2+ coordinated with peptide bond in BSA [20] and DOX was also loaded into the BSA structure due to its relative hydrophobic cavity. When DOX@BSA-Cu nanoparticles efficiently internalized into cancer cells, the overexpressed GSH could convert Cu2+ to Cu+, accompanying with the release of DOX. DOX could promote the production of H2O2, which was subsequently transformed to more toxic ·OH and efficiently induce cell death. To our knowledge, this is the first time low-dose DOX was used as supplement for H2O2 in CDT. The platform of DOX@BSA-Cu NPs achieving more effective CDT for cancer treatment demonstrates potentiality for future clinic research.
BSA was used as the drug carrier due to its good biocompatibility and modifiable groups [21-23]. The DOX@BSA-Cu NPs could be easily prepared through two steps at room temperature (Scheme 1). BSA-Cu was firstly obtained via Cu2+ coordination with BSA peptides, followed by DOX loading [20]. Both DOX@BSACu and BSA-Cu NPs showed uniform spherical morphologies with an average diameter of 30 nm in TEM images (Figs. S1 and S2 in Supporting information). BSA-Cu and DOX@BSA-Cu NPs were welldispersed in water with a narrow particle size distribution (Fig. 1a). Besides, the size of the NPs showed no significant difference before and after loading DOX, suggesting that drug loading did not obstruct the formation of the NPs. BSA and DOX@BSA NPs with spherical structures were also synthesized as compared with an average diameter of ~20 nm in TEM images (Fig. S1).

|
Download:
|
| Scheme 1. (a) The scheme of synthetic procedure for DOX@BSA-Cu NPs. (b) Schematic diagram of DOX@BSA-Cu NPs for enhanced chemodynamic therapy via self-generation of H2O2 in cancer cells. | |
{kind=link}

|
Download:
|
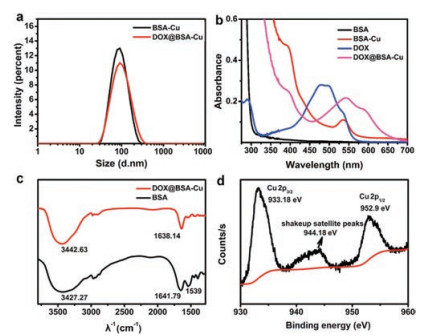
| Fig. 1. (a) DLS characterization of BSA-Cu and DOX@BSA-Cu NPs. (b) UV-vis absorption spectra of BSA (black line), BSA-Cu (red line), free DOX solution (blue line) and DOX@BSA-Cu (pink line) at room temperature. (c) FT-IR spectra of BSA and DOX@BSA-Cu. (d) Cu 2p XPS spectrum of the DOX@BSA-Cu NPs, the position of the satellite peak was pointed. | |
{kind=link}
Both the zeta potentials of DOX@BSA-Cu and BSA-Cu exhibited negative potential values (-33.5 and -31.5 mV) due to the negative charge of BSA, demonstrating that DOX was loaded within the NPs rather than adsorbing on their surface (Fig. S3a in Supporting information). The successful loading of DOX was confirmed by UV-vis absorption spectra (Fig. 1b). Pure BSA (black line) displayed only one absorption band centered at 280 nm. Compared with BSA, BSA-Cu (black line) and DOX@BSA-Cu (pink line) showed an absorption peak at 540 nm, which was the characteristic absorption peak of the complexes of Cu(II) and protein. Meanwhile, free DOX displayed main absorption peaks centered at 484 nm and 535 nm, which red shifted to 540 and 595 nm, respectively after loading into BSA. These changes in absorption may result from the interaction between DOX and Cu2+, affecting the electron transition in DOX. The DOX loading content was measured by using UV-vis spectroscopy. To avoid the influence caused by the red-shift of DOX absorption in DOX@BSA-Cu NPs, they were digested via trypsin before the measurement. The loading content of DOX was 4.8% with a ~95% loading efficiency, which is lower than other protein nanocarriers (~12% in loading content) [23-25], but it is enough for generating H2O2 rather than chemotherapy.
Fourier transform infrared spectroscopy (FT-IR) was performed to prove the combination between Cu2+ and BSA. The peaks at 3442, 1638 and 1539 cm-1 in were attributed to -OH, amide I (νC=O), and amide II (νC-N, δN-H) in BSA, respectively. Obviously, these peaks belonging to amide II (νC-N, δN-H) were weakened after interaction with Cu2+ in DOX@BSA-Cu NPs (Fig. 1c). The Cu doping amount was nearly 11% in DOX@BSA-Cu, calculated via ICP-MS analysis. It is noteworthy that the Cu content in the nanoparticles cannot induce toxic side effects [9, 11]. In fact, Cu ions with a certain concentration can be found in living systems. Further, X-ray photoelectron spectroscopy (XPS) showed that the spectrum peaked at 952.9 eV and 933.18 eV were assigned to Cu 2p1/2 and Cu 2p3/2 and shakeup satellite peaks can be clearly observed, which suggested that Cu2+ exists in DOX@BSA-Cu (Fig. 1d).
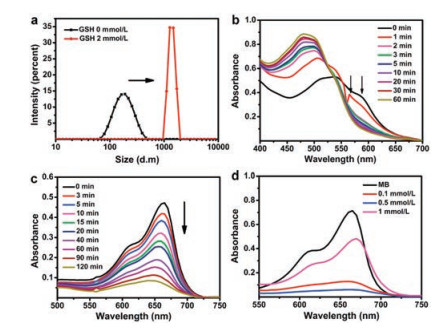
Next, we applied XPS to demonstrate that Cu2+ would be reduced to Cu+ by excess intracellular GSH after cell uptake of DOX@BSA-Cu. Compared with DOX@BSA-Cu, the peaks belong to Cu 2p3/2 and Cu 2p1/2 displayed almost no change. However, the original satellite peak at 944 eV (arrow points in Fig. 1d) vanished, which clearly proved that Cu2+ was reduced to Cu+ after reacting with GSH (Fig. S3b in Supporting information). It also demonstrated that DOX@BSA-Cu would consume GSH, avoiding ·OH being depleted by abundant GSH in cancer cells, which is beneficial for CDT. Additionally, the average particle size of NPs dramatically increased from 100 nm to mm size in response to GSH (2 mmol/L), suggesting that GSH reduced Cu2+ which unstabilized the NPs (Fig. 2a).

|
Download:
|
| Fig. 2. (a) The average diameter of DOX@BSA-Cu NPs before and after reaction with GSH (2 mmol/L), determined by DLS. (b) UV-vis absorption spectra of DOX@BSA-Cu after reaction with GSH (1 mmol/L). (c) The absorption changes of MB at different time points in solution containing NPs (50 μg/mL), GSH (0.1 mmol/L) and H2O2 (25 mmol/L). (d) The absorption changes of MB in response to different GSH concentrations in the presence of H2O2 (20 mmol/L). Each curve was measured after reaction for 2 h (CNPs = 50 μg/mL). (c) and (d), NPs were pre-treated with GSH for 15 min at 37 ℃, then reacted with H2O2. | |
{kind=link}
The real-time absorption spectrum changes of DOX@BSA and DOX@BSA-Cu were also monitored after incubation with GSH (1 mmol/L), respectively. As shown in Fig. 2b, the absorption of DOX@BSA-Cu gradually changed after incubation with GSH for different time periods, and was finally the same to that of free DOX, suggesting that DOX was released in the presence of GSH due to the dissociation of DOX@BSA-Cu. In contrast, the absorption of DOX@BSA kept constant even after incubating with GSH (1 mmol/L) for 1 h (Fig. S4a in Supporting information). Next, we studied the DOX release from DOX@BSA-Cu NPs treated with or without GSH. Obviously, the nanoparticles were dissociated in response to GSH, and more DOX (~60%) was released from NPs compared with nanoparticles without any treatment (~15%) (Figs. S2b and S4b in Supporting information). In addition, the release of Cu ion almost reached 60% after 1 h incubation with GSH (Fig. S4c in Supporting information). The results further proved that GSH resulted in the reduction of Cu2+, and subsequently destructed the DOX@BSA-Cu nanostructures.
H2O2 can be converted to more toxic ·OH through a Cu+ involved Fenton-like reaction. To prove the ability of NPs to generate ·OH, methylene blue (MB), a specific indicator for ·OH was used [26, 27]. MB displays intense absorption (664 nm) which can be bleached in response to ·OH. The typical absorption of MB gradually decreased after incubation of DOX@BSA-Cu in solution containing H2O2 and GSH, indicating that NPs demonstrated good ·OH production ability (Fig. 2c). Other groups including H2O2 + GSH, BSA-Cu + GSH, DOX@BSA-Cu + GSH did not result in degradation of MB. In contrast, in the presence of H2O2 and GSH, both BSA-Cu and DOX@BSA-Cu efficiently degraded MB (Fig. S5a in Supporting information). It is noteworthy that DOX can induce H2O2 only in cells, thus there was no obvious difference in the production of ·OH between BSA-Cu and DOX@BSA-Cu in solution. Further, the electron spin-resonance spectroscopy (ESR) using 5, 5-dimethyl- 1-pyrroline-N-oxide (DMPO) as the ·OH trapping agent was carried out. The specific signal further suggested that ·OH was generated by DOX@BSA-Cu in the solution containing GSH and H2O2 (Fig. S5b in Supporting information).
Next, we investigated the effect of GSH concentration on the production of ·OH. Cu2+ in the DOX@BSA-Cu would reduce the concentration of GSH which is beneficial for effective treatment. Clearly, MB experienced more degradation in responsive to 0.5 mmol/L GSH compared with 0.1 mmol/L GSH, because more Cu+ was produced which converted H2O2 to ·OH with the increase concentration of GSH. Moreover, further increasing the GSH concentration to 1 mmol/L, MB still could be degraded even the excessive GSH would be a scavenge for removing ROS in turn (Fig. 2d). These results demonstrated that DOX@BSA-Cu can produce ·OH efficiently after Cu2+ was reduced to Cu+ by GSH.
Confocal laser scanning microscopy (CLSM) was used to investigate the intracellular internalization of DOX@BSA-Cu (Fig. S6 in Supporting information). Red fluorescence of DOX was observed in cell plasma after 15 min incubation and the fluorescence intensity got to saturation after 1 h, indicating the nanoparticles can rapidly internalize into cancer cells due to their small size. We further examined the influence of BSA-Cu and DOX@BSA-Cu on the intracellular ROS by using 70-dichlorodihy-drofluorescein diacetate (DCHF-DA) as the ROS probe. DOX loaded in the NPs was expected to increase the H2O2 production [18, 19]. After the cells were treated with 100mg/mL of DOX@BSA-Cu NPs for 4 h, the ROS stress level in the cancer cells significantly increased (Fig. 3a, Fig. S7 in Supporting information). The discrepant ROS generation was observed in MCF-7 and 4T1 cells, because the self-generated H2O2 in the 4T1 cells is higher than that of MCF-7 cells [28]. Thus more ·OH was generated, and the ROS stress level was slightly increased in 4T1 cells. In contrast, the cells treated with BSA-Cu only demonstrated slight increase of ROS level. These results supported the hypothesis that DOX@BSA-Cu can enhance the generation of ·OH due to the presence DOX. DOX@BSA-Cu was expected to demonstrate more efficient CDT effect for cancer treatment.

|
Download:
|
| Fig. 3. (a) ROS staining in MCF-7 cells after treatment with 100mg/mL DOX@BSACu or BSA-Cu NPs for 4 h. Scale bars: 15mm. Cell viability of (b) 4T1 cells and (c) MCF-7 cells treated with different concentrations of BSA, DOX@BSA, BSA-Cu, DOX@BSA-Cu NPs for 24 h. | |
{kind=link}
The toxic effects of different nanoparticles to cancer cells were evaluated by methylthiazolyltetrazolium (MTT) assay. 4T1 and MCF-7 cells were incubated with different concentrations of BSA, DOX@BSA, BSA-Cu and DOX@BSA-Cu, respectively (Figs. 3b and c). BSA displayed no obvious cytotoxicity to cells as expected because of its excellent biocompatibility. DOX@BSA also showed negligible toxicity due to the low content of DOX. On the contrary, the viability of cells decreased gradually with increased the concentration of BSA-Cu and DOX@BSA-Cu, because toxic ·OH was generated intracellularly. In addition, DOX@BSA-Cu displayed higher inhibition rate than that of BSA-Cu. The cell viability inhibition rate was about 60% for both 4T1 and MCF-7 cells after incubation with 250 μg/mL DOX@BSA-Cu. However, at the same concentration, BSA-Cu resulted in 40% cell viability inhibition towards 4T1 and MCF-7 cells. The difference in cell toxicity was due to the presence of DOX which enhanced the generation of H2O2 and thus resulted in more efficient CDT. We also tested the cytotoxicity of the DOX@BSA-Cu NPs to normal cells. In contrast, DOX@BSA-Cu NPs demonstrated a relatively lower cytotoxicity toward COS-7 cells, due to the lower concentrations of GSH in normal cells. After 24 h incubation, the cell viability of COS-7 cells was still higher than 75%, even when a high concentration of DOX@BSA-Cu NPs was used (300 μg/mL) (Fig. S8 in Supporting information). Thus, DOX@BSA-Cu NPs demonstrated much higher cytotoxicity toward cancer cells than normal cells.
Furthermore, we adopted annexin V (AV)-FITC reagents to distinguish cell apoptosis stages after incubation with these nanoparticles. AV staining demonstrates either cell apoptotic or necrotic processes and PI stains the nucleus of late apoptotic or dead cells [29]. Negligible green and red fluorescence were observed after 4T1 cells were incubated with BSA and DOX@BSA nanoparticles (100 μg/mL) for 8 h due to biocompatibility of BSA and low-dose DOX (Fig. 4). However, obvious green fluorescence was observed from BSA-Cu treated cells, suggesting BSA-Cu resulted in the early apoptosis of most cancer cells. Notably, both strong green and red fluorescence could be detected from the cells after treatment with DOX@BSA-Cu, suggesting cells were in the late apoptosis.

|
Download:
|
| Fig. 4. Cell apoptosis test of 4T1 cells after treatment with 100 μg/mL BSA, DOX@BSA, BSA-Cu and DOX@BSA-Cu NPs for 8h. Single AV (green signals, λex=488nm, λem =525±15 nm) marked early apoptosis. Dual-stained cells with PI (red signals, λex = 488 nm, λem = 615±15nm) marked late apoptosis. Scale bars: 30μm. | |
{kind=link}
In summary, we successfully fabricated protein NPs (DOX@BSACu) for enhanced CDT by self-generating H2O2. This system consists of BSA coordinated with Cu2+, which would be reduced to Cu+ by GSH for catalyzing H2O2 in cancer cells. In addition, DOX was also released accompanying with the degradation of DOX@BSA-Cu. Different from traditional drug delivery system, where DOX was always used as a chemotherapeutics, DOX here with low-dose acted as a H2O2 generator. Thus, DOX@BSA-Cu demonstrated enhanced H2O2 generation compared with BSA-Cu. DOX@BSA-Cu showed efficient cell viability inhibition effect to cancer cells. We believe this work will further inspire the development of more efficient nanoplatforms for enhanced CDT for cancer treatment. In vivo experiments of the nanoparticles in CDT of cancer will be conducted in future after extensive evaluation of the safety and the stability of the nanoparticles in the blood circulation.
Declaration of competing interestThe authors declare that there are no conflicts of interest.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21808028, 21925802, 21878039, 21421005), and NSFC-Liaoning United Fund (Nos. U1608222, U1908202).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.031.
| [1] |
F. Bray, J. Ferlay, I. Soerjomataram, et al., CA Cancer J. Clin. 68 (2018) 394-424. DOI:10.3322/caac.21492 |
| [2] |
E.A. Kuczynski, D.J. Sargent, A. Grothey, et al., Nat. Rev. Clin. Oncol. 10 (2013) 571-587. DOI:10.1038/nrclinonc.2013.158 |
| [3] |
C. Holohan, S. van Schaeybroeck, D.B. Longley, et al., Nat. Rev. Cancer 13 (2013) 714-726. DOI:10.1038/nrc3599 |
| [4] |
S.Y. Li, H. Cheng, B.R. Xie, et al., ACS Nano 11 (2017) 7006-7018. DOI:10.1021/acsnano.7b02533 |
| [5] |
W.L. Liu, T. Liu, M.Z. Zou, et al., Adv. Mater. 30 (2018) 1802006. DOI:10.1002/adma.201802006 |
| [6] |
X.H. Huang, I.H. El-Sayed, W. Qian, et al., J. Am. Chem. Soc. 128 (2006) 2115-2120. DOI:10.1021/ja057254a |
| [7] |
L. Dykmana, N. Khlebtsov, Chem. Soc. Rev. 41 (2012) 2256-2282. DOI:10.1039/C1CS15166E |
| [8] |
C. Zhang, W. Bu, D. Ni, et al., Angew. Chem. 128 (2016) 2141-2146. DOI:10.1002/ange.201510031 |
| [9] |
C.H. Liu, D.D. Wang, S.Y. Zhang, et al., ACS Nano 13 (2019) 4267-4277. DOI:10.1021/acsnano.8b09387 |
| [10] |
Z. Zhou, J. Song, L. Nie, et al., Chem. Soc. Rev. 45 (2016) 6597-6626. DOI:10.1039/C6CS00271D |
| [11] |
B.J. Ma, S. Wang, F. Liu, et al., J. Am. Chem. Soc. 141 (2019) 849-857. DOI:10.1021/jacs.8b08714 |
| [12] |
L.S. Lin, J. Song, L. Song, et al., Angew. Chem. Int. Ed. 57 (2018) 4902-4906. DOI:10.1002/anie.201712027 |
| [13] |
D.W. Zheng, Q. Lei, J.Y. Zhu, et al., Nano Lett. 17 (2017) 284-291. DOI:10.1021/acs.nanolett.6b04060 |
| [14] |
Z.M. Tang, Y.Y. Liu, M.Y. He, et al., Angew. Chem. Int. Ed. 57 (2018) 2-13. DOI:10.1002/anie.201712504 |
| [15] |
J.H. Liao, H.R. Zheng, R. Hu, et al., J. Biomed. Nanotechnol. 14 (2018) 496-509. DOI:10.1166/jbn.2018.2510 |
| [16] |
Z.H. Yu, Y.C. Guo, H. Dai, et al., Mater. Express. 9 (2019) 467-474. DOI:10.1166/mex.2019.1525 |
| [17] |
S. Ayla, I. Seckin, G. Tanriverdi, et al., Int. J. Biochem. Cell. B 8 (2011) 390238. |
| [18] |
T. Simunek, S. Martin, O. Popelova, et al., Pharmacol. Rep. 61 (2009) 154-171. DOI:10.1016/S1734-1140(09)70018-0 |
| [19] |
P. Menna, S. Recalcati, G. Cairo, et al., Cardiovasc. Toxicol. 7 (2007) 80-85. DOI:10.1007/s12012-007-0011-7 |
| [20] |
F.P. Gao, P.J. Cai, W.J. Yang, et al., ACS Nano 9 (2015) 4976-4986. DOI:10.1021/nn507130k |
| [21] |
Q.Y. Yang, M.L. Wang, Y.B. Sun, et al., Chin. Chem. Lett. 30 (2019) 1224-1228. DOI:10.1016/j.cclet.2019.03.042 |
| [22] |
Y.Z. Wang, Y.J. Song, G.X. Zhu, et al., Chin. Chem. Lett. 29 (2018) 1685-1688. DOI:10.1016/j.cclet.2017.12.004 |
| [23] |
H. Hao, Q.M. Ma, F. He, et al., J. Mater. Chem. B 2 (2014) 7978-7987. DOI:10.1039/C4TB01359J |
| [24] |
J. Zhang, D. Zhang, Q. Li, et al., ACS Appl. Mater. Interfaces 11 (2019) 42904-42916. DOI:10.1021/acsami.9b13556 |
| [25] |
Y. Feng, R. Liu, L.C. Zhang, et al., ACS Appl. Mater. Interfaces 11 (2019) 44978-44988. DOI:10.1021/acsami.9b18124 |
| [26] |
S.H. Cao, J.L. Fan, W. Sun, et al., Chem. Commun. 55 (2019) 12956-12959. DOI:10.1039/C9CC06040E |
| [27] |
X.H. Zhang, Z.B. Geng, J. Jian, et al., Catalysts 10 (2020) 293-304. DOI:10.3390/catal10030293 |
| [28] |
H.J. An, C.H. Guo, D.D. Li, et al., ACS Appl. Mater. Interfaces 12 (2019) 17230-17243. |
| [29] |
M. Xiao, W. Sun, J.L. Fan, et al., Adv. Funct. Mater. 28 (2018) 1805128. DOI:10.1002/adfm.201805128 |