 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Nitrile is an important class of organic skeletons existed in many pharmaceuticals and natural products [1, 2]. Additionally, the nitrile group serves as versatile synthetic building motif in organic chemistry as it can be easily transformed into other important functional groups via routine manipulation [3]. Therefore, chemists have long-term interest in the development of practical methods for the synthesis of nitriles. Typical strategies include dehydration of aldoximes [4-8] or amides [9], dehydrogenation of amines [10-12], and cyanation of alkyl halides [13]. Recently, the iminyl radical-triggered C—C bond cleavage of cycloketone oximes has been successfully applied in the synthesis of diverse nitriles under cyanide-free conditions. The first example for the synthesis of nitriles from cycloketone oximes traced back to the year of 1991, Zard et al. developed the use of carboxymethyl oximes and cyclobutanone sulfenylimines as iminyl radical precursors to prepare nitriles [14]. Later, many organic chemists reported the similar ring-opening reaction of cycloketone oximes [15-17]. Among this, photocatalysis is a convenient tool and widely used in organic synthesis due to its mild reaction conditions, availability and high efficiency [18-20]. Regarding the importance of nitriles and the efficiency of photocatalysis, recent development for the synthesis of nitriles via photoinduced C—C bond cleavage of cycloketone oximes is summarized in this paper, including photocatalysis and the combination of photocatalysis and metal catalysis.
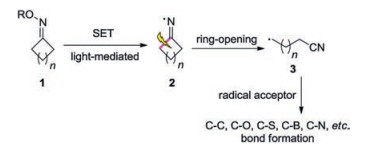
According to the literature, the cycloketone oximes 1 can form iminyl radical 2 through single electron transfer (SET) under photocatalysis, which would undergo C—C cleavage to generate relatively active cyanoalkyl radical intermediate 3. The cyanoalkyl radical 3 can be trapped by radical acceptors to form various bonds. Herein, we summarize the recent advances for the synthesis of nitriles through photoinduced C—C bond cleavage of cycloketone oximes classified by the type of C—X bond formation. Various compounds possessing nitriles can be efficiently accessed via this method (Scheme 1).

|
Download:
|
| Scheme 1. Synthesis of nitriles from cycloketone oximes according to the type of bond formation. | |
{kind=link}
2. C—C bond formation 2.1. Alkene as the radical acceptor through reductive initiation
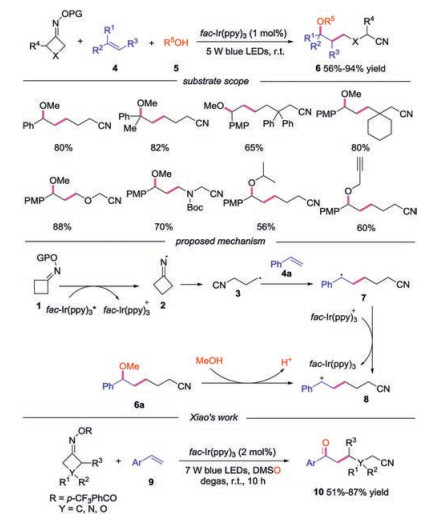
In 2017, Zhou's group reported a three-component cyanopropylation/etherification of unactivated alkenes through a photoredox approach [21]. As for the substrate scope, O-acyl oximes with various styrenes in MeOH furnished the corresponding products in good yields. Additionally, oximes with different functional groups all worked well, affording the desired products in good to excellent yields. Moreover, proparyl alcohol underwent reactions smoothly, though with moderate reactivity. A plausible mechanism was proposed, as shown in Scheme 2. The reactions started with the visible-light-driven SET reduction of O-acyl oxime 1 by the photoexcited catalyst fac-Ir(ppy)3* to generate the iminyl radical 2 along with the oxidized photocatalyst. The iminyl radical 2 then underwent a ring-opening by C—C cleavage to form cyanoalkyl radical 3. Addition of radical 3 to alkene 4a produced carbon radical 7, which was oxidized to carbocation intermediate 8 by fac-Ir(ppy)3+ with the regeneration of photocatalyst. Finally, the carboncation 8 went through nucleophilic reaction with MeOH to generate the final product 6a. Except the final nucleophilic attack by MeOH, the carbocation 8 could also be oxidized by DMSO to form the ketonitriles, which was developed by Xiao and co-workers in 2018 [22].

|
Download:
|
| Scheme 2. Three-component reaction for the synthesis of alkylnitriles and ketonitriles. | |
{kind=link}
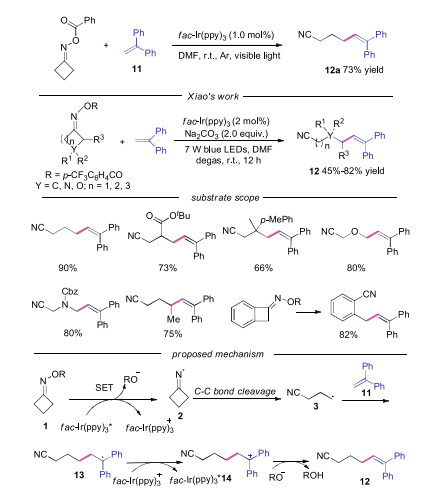
Excepted reacting with nucleophiles, the carbocation could also went through an elimination process to provide alkene products. In 2017, Shi's group reported the copper catalyzed coupling reaction of cycloketone oximes with unsaturated compounds 11. In this work, they also discovered that the coupling reaction could proceed successfully under photocatalysis conditions, providing the cyano-containing product 12a in good yield [23]. In 2018, Xiao's group developed the similar visible-light-driven iminyl radical-mediated C—C bond cleavage/radical addition cascade reaction of oxime esters 1 with unsaturated compounds 11 in the presence of base [24]. Various C(sp3)-C(sp2) bonds could be constructed through this redox-neutral process. Symmetric monosubstituted O-acyl oximes bearing functional groups at 3-position and disubstituted substrates proceeded smoothly to give the desired products. Furthermore, both oxetan-3-one and 1-Cbz-3-azetidinone derived O-acyl oximes participated in the reaction well. Functional groups at the 2-position of O-acyl oximes also demonstrated good reaction efficiency. Once again, benzocyclobutenone-derived substrate proved to be suitable for the reaction. The authors proposed a mechanism shown in Scheme 3. The carbocation intermediate 14 was generated through the known process, which went through base-mediated deprotonation to afford the alkene product 12.

|
Download:
|
| Scheme 3. Visible-light driven cascade reaction of cycloketone oxime esters and alkenes. | |
{kind=link}
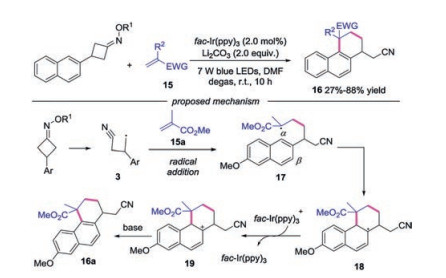
When cyanoalkyl radical 3 added to the electron-deficient alkene, the generated radical could attack the aromatic ring to produce the cycloadduct. In 2018, Xiao's group developed a photoredox-catalyzed radical addition/cyclization cascade to generate cyanoalkylated 1, 2, 3, 4-tetrahydrophenanthrenes with good yields and excellent regioselectivity [25]. The authors proposed a mechanism (Scheme 4), showing that the radical addition would occur between cyanoalkyl radical 3 and electrondeficient alkene 15 leading to radical intermediate 17. Then the radical 18 was afforded by the intramolecular radical addition to the α-site of naphthalene. Subsequently, the radical 18 was oxidized by fac-Ir(ppy)3+ to give rise to carbocation 19, which was deprotonated under base conditions to produce the final product 16.

|
Download:
|
| Scheme 4. Synthesis of tetrahydrophenanthrenes through photoinduced C—C bond cleavage of cycloketone oximes. | |
{kind=link}
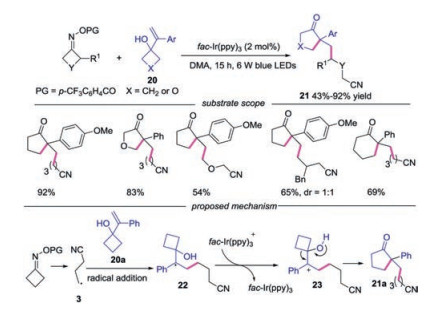
In the same year, Xiao's group also employed this methodology in the reaction with alkenylcyclobutanols to prepare cyclic ketones [26]. A variety of substrates were found to be compatible under the conditions. The 3-(1-phenylvinyl)oxetan-3-ol, symmetric monosubstituted O-acyl oximes and unsymmetrical cyclobutanonederived O-acyl oximes were suitable in this transformation. Furthermore, 1-(1-phenylvinyl)-cyclopentanol was also tolerated, affording the desired product in good yields. The plausible mechanism was shown in Scheme 5. Cyanoalkyl radical 3 reacted with 1-(1-phenylvinyl)cyclobutanol 20a leading to radical 22, which was oxidized to carbocation 23 by fac-Ir(ppy)3+. Finally, the carbocation 23 underwent alkyl migration and deprotonation to provide the product 21a.

|
Download:
|
| Scheme 5. Visible-light induced synthesis of cyclic ketones from cycloketone oximes with cyclopentanol. | |
{kind=link}
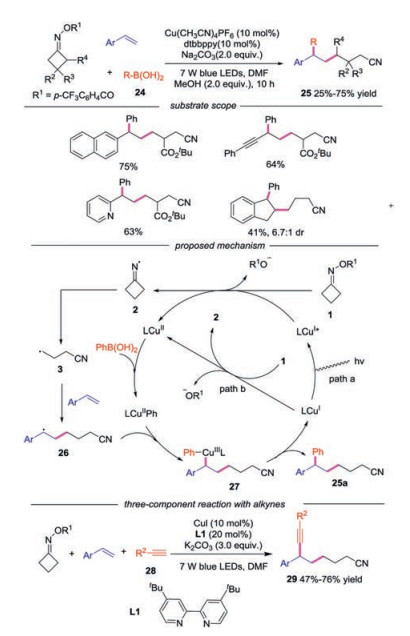
A copper-catalyzed, visible-light-driven three-component radical cross-coupling of oxime esters, boronic acids and styrenes was developed as well [27]. A wide range of boronic acids, alkenes and oxime esters were compatible in this reaction. A CuⅠ/CuⅡ/CuⅢ-based catalytic cycle was proposed during the transformation. The reaction started with SET reduction of oxime esters 1 by either photoexcited LCuⅠ* or the ground-state LCuⅠ giving rise to iminyl radical 2, which underwent C—C bond scission to provide cyanoalkyl radical 3. Then, the radical 3 was captured by alkene to form radical 26. Meanwhile, the LCuⅡ complex underwent transmetallation with boronic acid 24 to provide LCuⅡPh species. Subsequently, LCuⅡPh was oxidized by radical 3 to generate the high-valence CuⅢ complex and the final product 25 was afforded through reductive elimination. In 2019, Chen and co-workers reported a three-component cross-coupling reaction of cycloketone oxime esters, alkene and terminal alkynes, providing cyanoalkyl-containing propargylic compounds in general good yields [28]. The reactions proceeded with similar mechanism shown in Scheme 6.

|
Download:
|
| Scheme 6. Visible-light induced three-component reaction with cycloketone oxime esters, alkenes and boronic acids/alkynes. | |
{kind=link}
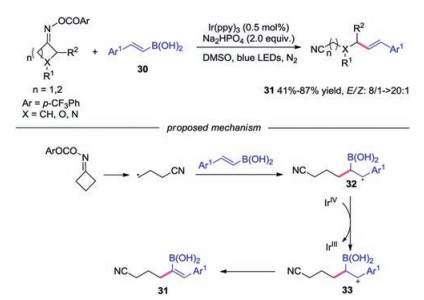
In 2018, Yu's group reported the photocatalyzed remote C—C vinylation of cycloketone oximes with vinyl boronic acids [29]. Various γ-vinyl nitriles were efficiently synthesized through this method. The authors proposed mechanism in Scheme 7. The cyanoalkyl radical 3 added to vinyl boronic acids 30 to provide the radical intermediate 32. Subsequently, the radical 32 was oxidized by IrⅣ to produce carboncation 33. Finally, deprotonation afforded the desired γ-vinyl nitriles 31.

|
Download:
|
| Scheme 7. Visible-light induced synthesis of γ-vinyl nitriles from cycloketone oximes with vinyl boronic acids. | |
{kind=link}
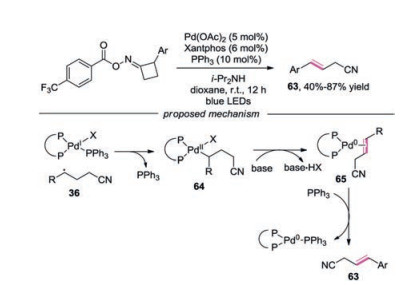
The synthesis of β-aroyl hexanenitriles and (E)-4-arylbut-3-enenitriles through a photoexcited Pd complex activation of cyclobutanone oxime esters was reported by Fu and co-workers [30]. For the ring-opening addition reaction, functional groups such as sulfone on trimethyl((1-phenylvinyl)oxy)silane were compatible. Additionally, α-heteroparyl silyl enol ethers including pyridyl and furanyl participated in the addition reaction well. The mechanism proposed for this palladium-catalyzed ringopening addition of cyclobutanone oxime esters was presented in Scheme 8. The palladium complex would activate oxime ester through photoinduced SET to generate a hybrid iminyl radical PdⅠ intermediate 35, which underwent C—C cleavage to produce cyanoalkyl PdⅠ species 36. Radical 36 would then interact with silyl enol ether leading to a hybrid benzyl radical PdⅠ 37, which underwent benzyl association to provide benzyl-PdⅡ 38. After β-H elimination of 38, intermediate 39 would be formed, which would produce alkylated silyl enol ether with the assistance of PPh3. After hydrolysis, ketone product 34 would be afforded.

|
Download:
|
| Scheme 8. Photoexcited Pd0 complexinduced addition reactionof cycloketoneoximes. | |
{kind=link}
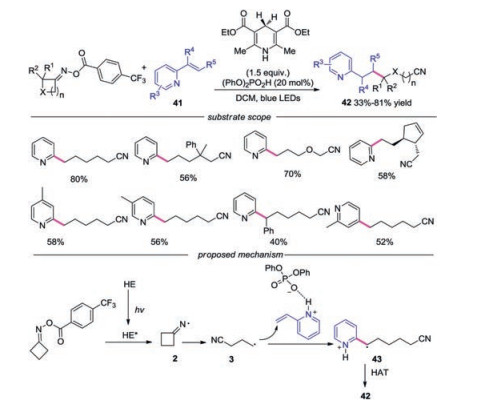
In 2020, Liu's group developed a visible-light-initiated iminyl radical-mediated C—C bond cleavage and functionalization of cycloketone oximes (Scheme 9) [31]. Regarding the substrates, various functional groups on the four-membered ring participated in the reaction well. 2-Alkenylpyridine and 4-vinylpyridine promoted the reaction smoothly. According to the mechanism studies, the reaction was initiated by HE* rather than an EDA complex. After a single electron transfer, cyanoalkyl radical 3 was generated through C—C bond cleavage, which was trapped by charged 2-vinylpyridine to produce alkyl radical 43. The final product was generated through abstraction of a hydrogen atom from HE·+ or HE.

|
Download:
|
| Scheme 9. Visible-light induced reaction of cycloketone oximes with HE as the reductant. | |
{kind=link}
2.2. Alkane as the radical acceptor through reductive initiation
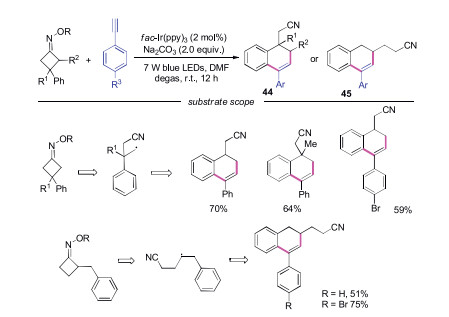
In 2018, Xiao's group described the reaction of cycloketone oximes with alkenes to generate cyano-containing alkenes (Scheme 3). Additionally, the cyclic products were produced when alkynes were employed (Scheme 10) [24]. A variety of 1, 2-dihydronaphthalenes 44 and 45 were efficiently obtained through this method. The reactions were proceeded through radical cascade processes.

|
Download:
|
| Scheme 10. Visible-light induced cycloaddtion reaction of cycloketone oximes and alkynes. | |
{kind=link}
2.3. Aromatic ring as the radical acceptor through reductive initiation
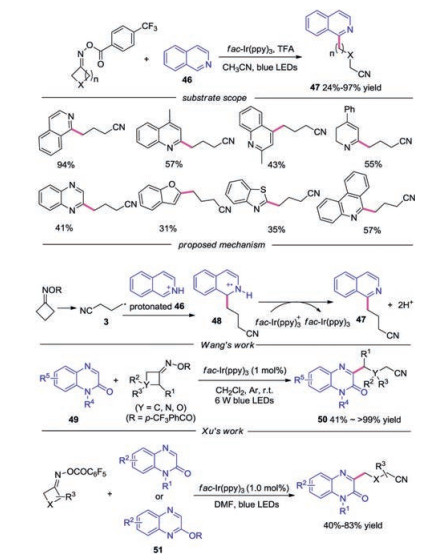
In 2019, the visible-light-driven Minisci-type C—H cyanoalkylation of heteroarene with cycloketone oximes was reported [32]. Various heterocyclic compounds were well tolerated in this reaction, such as isoquinolines, quinolones, quinoxalines, benzofurans, benzothiazoles and phenanthridins. A possible mechanism was proposed, as shown in Scheme 11. Radical 3 was captured by protonated isoquinoline to produce radical cation 48, which was oxidized by fac-Ir(ppy)3+ to afford the final product. In the same year, Wang's group [33], Xu and co-workers [34] reported a similar transformation with quinoxalinones as the radical acceptor.

|
Download:
|
| Scheme 11. Visible-light induced cyanoalkylation of heteroarene with cycloketone oxime esters. | |
{kind=link}
2.4. Metal species as the radical acceptor through reductive initiation
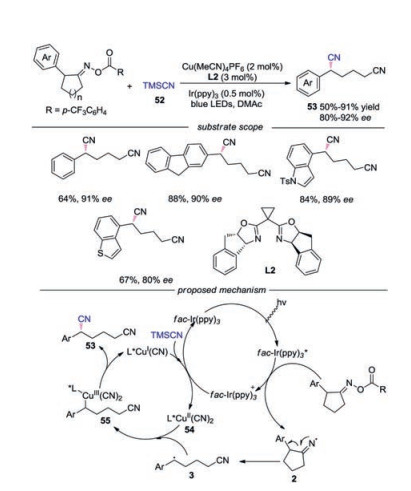
An enantioselective ring-opening cyanation reaction of cycloketone oxime esters to access chiral 1, 5- and 1, 6-dinitriles was described [35]. The reaction of cyclopentanone oxime esters required the dual catalyst of photo- and copper-catalysis while the reaction with cyclobutanone could be accessed by copper catalysis only. A broad substrate scope was demonstrated, and a series of cyclopentanone oxime esters with various substituted aromatic rings were well tolerated. A dual photoredox/copper catalysis mechanism for reaction of cyclopentanone oxime esters was proposed (Scheme 12). Initially, oxime ester was reduced by photoexcited fac-Ir(ppy)3* to generate iminyl radical 2. Meanwhile, the L*CuⅠCN was oxidized by fac-Ir(ppy)3+ species with participation of TMSCN to afford CuⅡ species 54. The benzylic radical 3, produced by the C—C bond cleavage of iminyl radical 2, was captured by L*CuⅡ(CN)2 to form CuⅢ species 55. Subsequently, reductive elimination of 55 would occur leading to the desired product.

|
Download:
|
| Scheme 12. Visible-light induced 1, 6-dinitriles reaction of cycloketone oximes. | |
{kind=link}
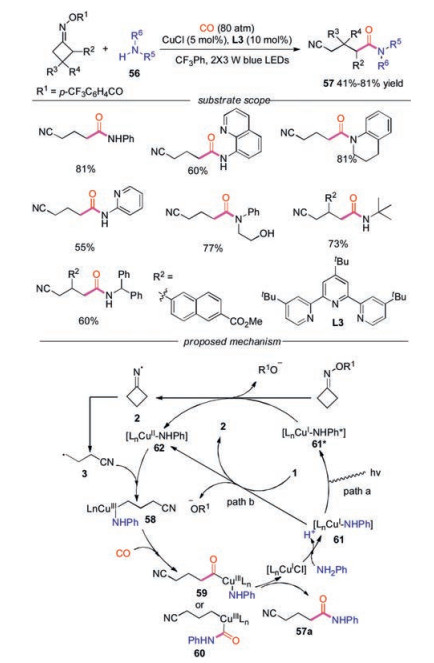
A visible light-induced, copper-catalyzed radical aminocarbonaylation of oxime esters with amines and CO gas without exogenous photosensitizer was developed by Xiao and co-workers [36]. This method exhibited broad substrate scope and highly functional group tolerance with both cycloketone oxime esters and alkyl/aryl amines. Aryl amines with fused aromatic group, cyclic or acylic secondary amines and amine-substituted pyridines participated in this reaction well. Moreover, sterically demanding primary amines were compatible under the conditions. A visible-light-driven CuⅠ/CuⅡ/CuⅢ-based catalytic cycle was proposed for this radical amino-carbonylation reaction (Scheme 13). The reaction would be initiated with the SET reduction of 1 by either the LnCuⅠ-NHPh* or the ground state LnCuⅠ-NHPh, affording an iminyl radical 2. Then radical 2 underwent C—C bond cleavage to produce cyanoalkyl radical 3, which was intercepted by CuⅡ complex to generate high-valence CuⅢ complex 58. CuⅢ complex 58 would then undergo coordination and insertion of CO to afford acylcopper intermediate 59 or 60, which provided the final product after reductive elimination.

|
Download:
|
| Scheme 13. Visible-light induced aminocarbonylation of cycloketone oximes. | |
{kind=link}
In 2019, Fu's group reported the photoexcited Pd0 species induced addition reaction of cycloketone oxime esters with silyl enol ethers (Scheme 8) [30]. Additionally, β-elimination reaction occurred with cycloketone oxime esters in the absence of silyl enol ethers, affording the corresponding alkene products in good yields. The proposed mechanism was shown in Scheme 14. The PdⅠ complex, generated by cycloketone and Pd0 complex (Scheme 8), associated with cyanoalkyl radical to generate PdⅡ complex 64 along with the release of PPh3. Next, β-elimination occurred under base conditions, and the final product was obtained after the diassociation with palladium.

|
Download:
|
| Scheme 14. Photoexcited Pd0 complex induced β-elimination reaction of cycloketone oximes. | |
{kind=link}
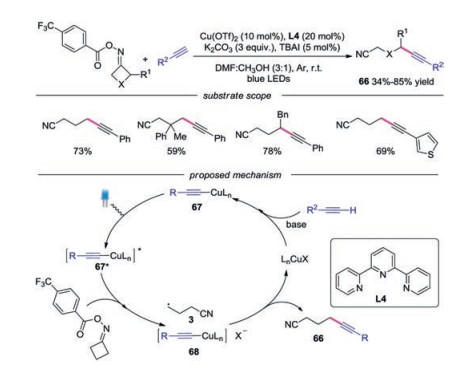
Recently, Shi and co-workers developed a novel transformation for the C(sp3)-C(sp) bond formation via photo-induced coppercatalyzed C—C bond cleavage of cycloketone oximes (Scheme 15) [37]. Various functionalized alkynyl nitriles were easily obtained with cycloketone oximes and terminal alkynes. The proposed mechanism showed that the copper acetylide complex 67, generated by alkyne and copper catalyst, was irradiated by visible light to afford photoexcited complex 67*. Subsequently, the cycloketone oxime esters were reducted by photoexcited complex 67* to produce 68 and cyanoalkyl radical 3. Finally, the cyanoalkyl radical 3 would be trapped by complex 68, providing the alkynyl nitrile along with the copper salt.

|
Download:
|
| Scheme 15. Visible-light induced copper-catalyzed C(sp3)-C(sp) bond formation from cycloketone oximes. | |
{kind=link}
2.5. Reactions through oxidative initiation
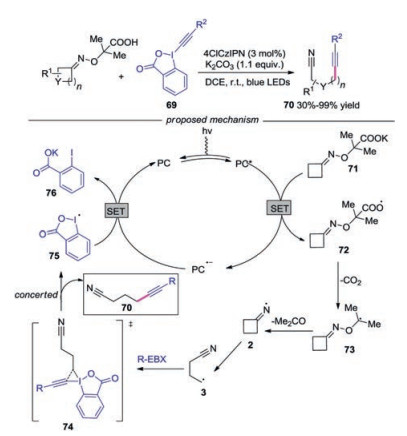
Waser's group described the synthesis of alkynyl nitriles with cycloketone oximes by organic dyes (Scheme 16) [38]. Various functional groups were well tolerated during the transformation. Mechanistic studies showed that the reaction was initiated with the oxidation of potassium carboxylate 71 by the excited state of organic dye to generate radical 72. Then, the fast decarboxylation gave rise to α-oxy radical 73, which would provide the iminyl radical 2 by N–O scission with the release of acetone. Afterwards, the iminyl radical 2 fragmented into alkyl nitrile radical 3, which reacted with EBX affording the desired alkynylnitrile 70 through 74.

|
Download:
|
| Scheme 16. Visible-light-induced synthesis of alkynyl nitriles from cycloketone oximes and EBX. | |
{kind=link}
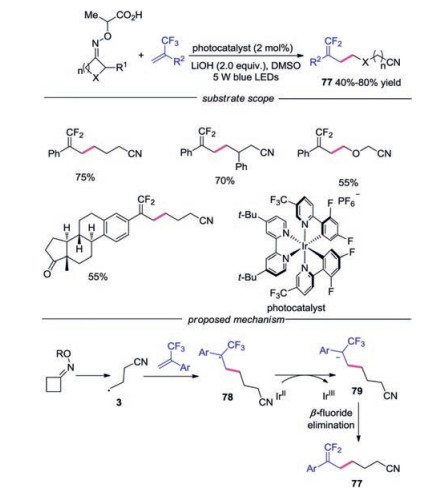
A visible-light-driven γ, γ-difluoroalkylation of cycloketone oximes esters with trifluoromethyl alkenes via C—C and C—F bond cleavage was developed, providing a series of cyanosubstituted gem-difluoroalkenes in good yields (Scheme 17) [39]. A wide range of substrates with sensitive functional groups were well tolerated in this reaction, including alkenes derived from estrone, symmetrical cyclobutanone oxime esters, oxime ethers derived from oxetan-3-one. As for the mechanism, the cyanoalkyl radical 3 was generated through similar pathway shown in Scheme 15. Subsequently, radical 3 would attack the double bond of α-CF3 alkene leading to α-CF3 alkyl radical 78. Reduction of α-CF3 alkyl radical 78 afforded carbanion 79, which would went through β-fluoride elimination to deliver the final cyano-substituted gemdifluoroalkene 77.

|
Download:
|
| Scheme 17. Visible-light-induced γ, γ-difluoroalkylation of cycloketone oximes esters. | |
{kind=link}
In 2019, Leonori's group described a dual photoredox/nickel strategy for remote functionalization with cycloketone oximes via iminyl radicals (Scheme 18) [40]. Aryl bromide, alkynes and alkyl bromide were well tolerated, enabling the development of distal arylation, vinylation and alkylation of nitrile-containing compounds. The proposed mechanism was shown in Scheme 17. The reaction started with the oxidation of oxime-carboxylate 71 by photoexcited photocatalysis to produce cyanoalkyl radical 3. Meanwhile, the Ni0 catalyst would undergo oxidative addition on an aryl bromide to produce NiⅡ species [41]. Then, the radical transmetallation between radical 3 and NiⅡ species delivered NiⅢ species 85, which went through reductive elimination to afford the final product 82.

|
Download:
|
| Scheme 18. Photo/nickel catalyzed remote functionalization with cycloketone oximes. | |
{kind=link}
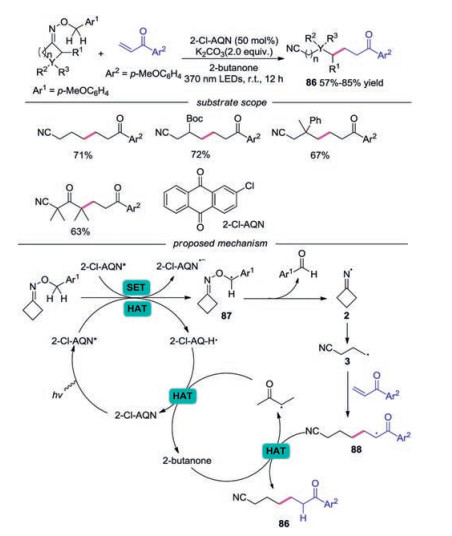
In 2019, a metal-free, light-driven and iminyl radical-mediated ring opening C—C bond cleavage/addition cascade reaction of O-4-methoxybenzyl oxime ethers with alkenes was described (Scheme 19) [42]. This reaction showed a broad substrate with excellent functional group tolerance, affording the corresponding oxo-nitriles in good yields. For example, monosubstituted and 3, 3-disubstituted O-benzyl oxime ethers promoted the reaction well. Additionally, oxetan-3-one derived oxime ether was well employed in this reaction. The proposed mechanism indicated that the reaction was initiated with the abstraction of hydrogen atom at the benzylic position of cycloketone oximes by photoexcited catalyst 2-Cl-AQN* to form benzylic radical 87, which underwent β-N—O bond scission to provide cyclic iminyl radical 2. Further β-C-C bond cleavage of iminyl radical 2 would give rise to cyanoalkyl radical 3, which would attack alkene subsequently to furnish radical 88. Followed by another HAT from solvent, the final product 86 would be produced.

|
Download:
|
| Scheme 19. Light-induced C—C bond forming reaction of cycloketone oxime ethers and alkenes. | |
{kind=link}
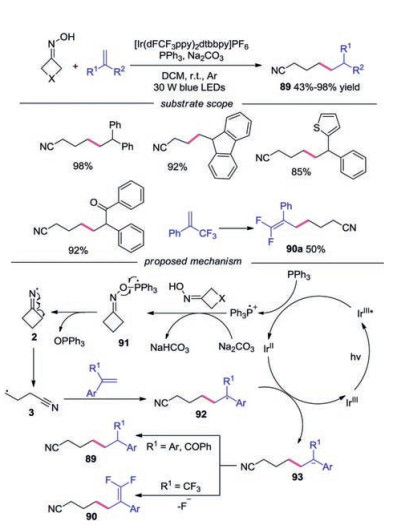
Yang and co-workers developed a photoinduced, phosphoranyl radical-mediated method for the direct N—O cleavage of cycloketone oximes through a polar/SET crossover process (Scheme 20) [43]. Alkenes with varied electronic and structural features participated in this reaction well. The fluorine analogue, olefins containing heterocycles and olefins with electron-withdrawing groups were well tolerated under the conditions. Moreover, β-flouride elimination occurred to afford gem-difluoroalkenes under the standard conditions when α-CF3 alkenes were employed. A possible mechanism was proposed, which showed that Ph3P was oxidized by photoexcited IrⅢ* to generate Ph3P·+. Subsequently, a polar/SET crossover process took place to furnish radical 91 in the presence of Na2CO3. Radical 91 would undergo N—O scission to generate the key iminyl radical 2, which would go through C—C bond cleavage giving rise to cyanoalkyl radical 3. Cyanoalkyl radical 3 was then captured by alkene to produce radical 92. The carbanion intermediate 93 was formed through reduction by IrⅡ. Protonation of intermediate 93 occurred with regular alkene to form alkyl nitrile, while β-flouride elimination of 93 afforded the gem-difluoroalkene in the case of α-CF3 alkene.

|
Download:
|
| Scheme 20. Photoinduced phosphoranyl radical-mediated C—C bond forming reaction of cycloketone oximes. | |
{kind=link}
2.6. Miscellaneous
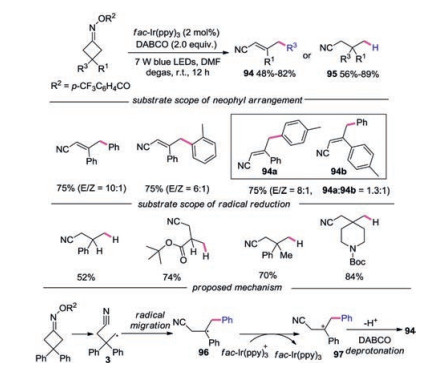
In 2018, Chen and co-workers reported the photocatalytic neophyl rearrangement and reduction of distal carbon radicals under visible light irradiation of O-acyl oximes (Scheme 21) [44]. Regarding for the substrate scope of neophyl rearrangement, symmetric disubstituted substrates with functional groups at the para-position worked well. When R3 and R1 were different substituted aryl groups, the selectivity of migration was dependent on their steric and electronic characters. Substrate with a methyl group at the para-position provided products 94a and 94b with moderate selectivity of aryl migration, while substrate with a methyl group at the ortho-position of one phenyl ring resulted in preferential migration. For the substrate scope of reduction of distal carbon radicals, the mono-substituted substrates at 3-position participated in the reaction well. Aryl and alkyl disubstituted substrates proceeded smoothly to furnish 3, 3-disubstituted propionitriles. The proposed mechanism showed that carbon radical 3 went through neophyl rearrangement to afford more stabilized secondary carbon radical 96. Subsequently, radical 96 was oxidized by fac-Ir(ppy)3 to form carboncation 97. Ultimately, DABCO promoted deprotonation of carboncation 97 would afford α, β-unsaturated nitrile 94.

|
Download:
|
| Scheme 21. Neophyl rearrangementand radical reduction reaction of cycloketone oximes. | |
{kind=link}
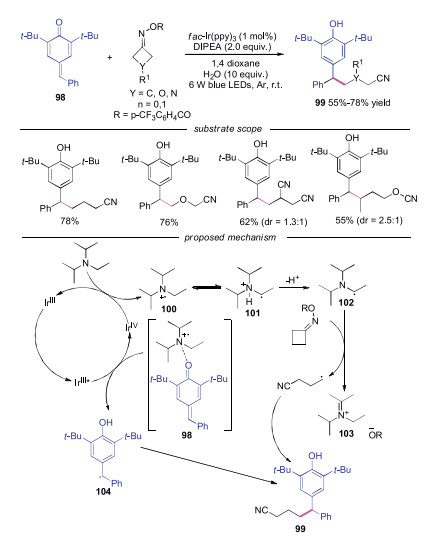
In 2019, Li and co-workers reported an efficient method to synthesize cyanoalkylated diarylmethanes, which were the key intermediates for the synthesis of GPR 40 agonists (Scheme 22) [45]. Bromoacetonitriles and cycloketone oxime esters were employed as the cyanoalkyl radical source. For the reactions in which cycloketone oxime esters acted as cyanoalkyl radical source, various substituted cyanoalkylated diarylmethanes 99 were efficient obtained in moderate to good yields. The authors proposed the mechanism in Scheme 22. The photoexcited IrⅢ* catalyst was oxidized by para-quinone methides 98 to generate IrⅣ catalyst and benzyl radical 104. Subsequently, α-amino radical 101 generated by DIPEA was oxidized by cycloketone oxime esters to afford the cyanoalkyl radical along with the iminium salt 103. Finally, the radical coupling between benzyl radical 104 and cyanoalkyl produced the desired cyanoalkylated diarylmethanes 99.

|
Download:
|
| Scheme 22. Visible-light induced synthesis of cyanoalkylated diarylmethanes from cycloketone oxime ketone and para-quinone methides. | |
{kind=link}
3. C—S, C—Se bond formation
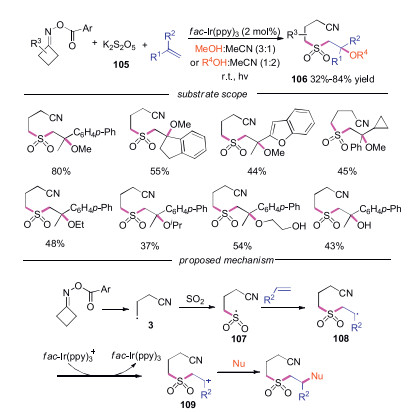
Organosulfur compounds existed widely in nature products and bioactive compounds. Therefore, the formation of C—S bond has gained great interest in recent years [46]. In 2019, Wu and coworkers reported a photoinduced multicomponent sulfonylation of O-acyl oximes through iminyl radicals with the insertion of sulfur dioxide (Scheme 23) [47]. Variation of alkenes, oximes and alcohols were compatible in this transformation. For example, alkenes with benzofuranyl and cyclopropane substitutions were well suitable. Moreover, ethane-1, 2-diol and other secondly alcohol participated the reaction smoothly. Additionally, various β-hydroxy sulfones were produced when the reactions were proceeded in water and MeCN. Mechanistic studies showed that the cyanoalkyl radical 3 was trapped by sulfur dioxide to form sulfony radical 107, which attacked the alkene to generate another radical intermediate 108. Subsequently, radical intermediate 108 was oxidized by photocatalyst to afford radical cation 109, which would react with nucleophile to form the desired product 106.

|
Download:
|
| Scheme 23. Photoinduced multi-component sulfonylation reaction of cycloketone oximes. | |
{kind=link}
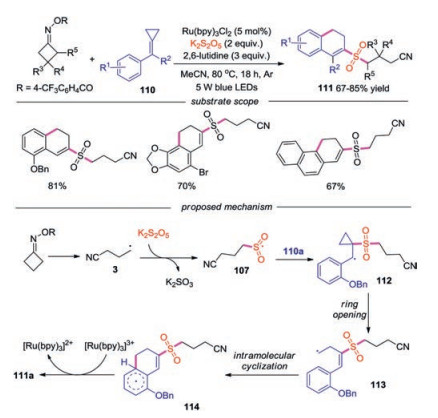
In 2020, Tang and co-workers reported a visible-light induced dual C—C bond cleavage of cycloketone oximes and methylenecyclipropanes (MCPs) for the synthesis of 2-cyanoalkylsulfonated 3, 4-dihydronaphthalenes via the insertion of sulfur dioxide (Scheme 24) [48]. Variation of substitution on cycloketone oxime esters and methylenecyclipropanes were well compatible under the conditions. For instance, reactions of halo-substituted MCPs and naphthyl-substituted MCP proceeded smoothly. The proposed mechanism revealed that the cyanoalkyl radical 3 captured sulfur dioxide to form cyanoalkylsulfonyl radical 107, which would attack the double bond in 110a leading to radical 112. Another cleavage of C—C bond would then occur to afford radical 113, which underwent intramolecular cyclization giving rise to radical 114. Followed by SET via [Ru(bpy)3]3+ and deprotonation by 2, 6-lutidine, the target product 111a would be afforded.

|
Download:
|
| Scheme 24. Photoinduced multi-component sulfonylation reaction of cycloketone oximes. | |
{kind=link}
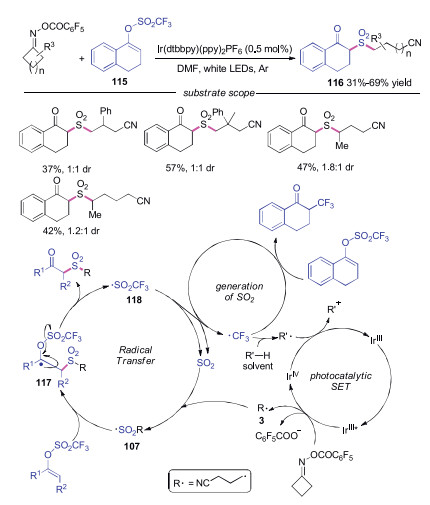
A photoredox-catalyzed in situ SO2-capture reaction was developed by Lu's group (Scheme 25) [49]. This reaction tolerated a variety of functional groups, and a range of desired products was afforded in good yields. The proposed mechanism showed that the cyanoalkyl radical 3 was generated through reduction of cycloketone oxime by photoexcited catalyst followed by C—C cleavage. Then, this cyanoalkyl radical 3 captured the in situ generated SO2 to form sulfonyl radical 107, which reacted with enol triflate 115 leading to intermediate 117. Elimination of radical intermediate 117 provided the product 116 with the release of trifluoromethylsulfonyl radical 118, which would regenerate SO2 through fragmentation along with the release of trifluoromethyl radical. The trifluoromethyl radical had two functions: it could participate in the decomposition of 115 to produce α-trifluoromethylated ketone as the side product. On the other hand, hydrogen abstraction from solvent (R'-H) would afford radical R'·, which could reduce IrⅣ to IrⅢ to complete the catalytic cycle.

|
Download:
|
| Scheme 25. Photoinduced SO2-capture reaction of cycloketone oximes. | |
{kind=link}
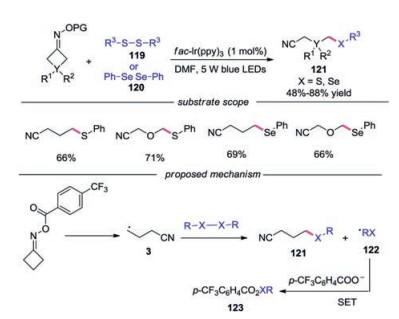
In 2019, Zhou's group reported the photocatalytic thiolation and selenylation of cycloketone oxime esters via iminyl radicaltriggered C—C bond cleavage, providing a unified approach to alkyl sulfur and selenium compounds tethered to nitrile group (Scheme 26) [50]. Again, oxime esters with various functional groups were well tolerated under the conditions, providing the desired products in generally good yields. A plausible mechanism for this C(sp3)-S, C(sp3)-Se bond forming reaction was proposed. It was reasoned that the cyanoalkyl radical 3 would be captured by disulfide or diselenide leading to C(sp3)-S or C(sp3)-Se bond respectively along with the formation of radical 122. The radical 122 was then oxidized by IrⅣ to complete the catalytic cycle.

|
Download:
|
| Scheme 26. Photoinduced C—S/Se bond formation reaction of cycloketone oximes. | |
{kind=link}
4. C—O bond formation
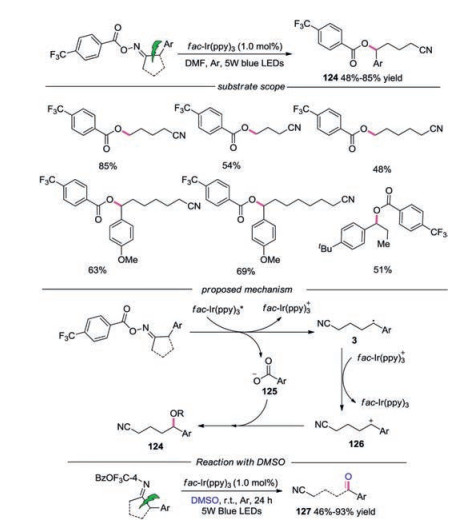
In 2018, Shi's group described a photoinduced protocol for the synthesis of cyano-containing benzoates by fragmentation-rearrangement sequence of cycloketoxime esters (Scheme 27) [51]. It was found that four-membered and less-strained substrates were tolerated in this transformation. Furthermore, the noncyclic oxime esters were also suitable, providing the products without cyano group. The proposed mechanism revealed that the reaction was initiated with the reduction of cycloketone oximes ester by photoexcited catalyst leading to cyano radical 3 with the release of carboxylate 125. Subsequently, cyanoalkyl radical 3 was oxidized by IrⅣ to afford cationic intermediate 126. Followed by the nucleophilic attack between carboxylate 125 and cationic intermediate 126, the final product 124 would be produced. Almost the same time, Shi and co-workers also reported a photoinduced strategy for the synthesis of cyano-containing ketones by C—C bond scission/oxidation sequence of cycloketone oximes [52]. In this reaction, the cation intermediate 126 was oxidized by DMSO to form the final ketone product 127.

|
Download:
|
| Scheme 27. Photoinduced fragmentation/rearrangement and oxidation reaction of cycloketone oximes. | |
{kind=link}
5. C—X (F, Cl) bond formation
In 2018, Leonori's group described the synthesis of a variety of differentially functionalized nitriles via a photoinduced cascade strategy (Scheme 28) [53]. These mild, visible light mediated protocols could be used for remote fluorination, chlorination and azidation, and the modification of structurally complex molecules and bioactive compounds. Oximes derived from cyclobutanes, cyclopentanones, cyclohexanones and even cycloheptanones were workable in this reaction, affording a broad range of fluorination and chlorination products in good yields. Mechanistic studies showed that after oxidation of oxime, radical 72 would be generated and underwent N—O bond cleavage to form iminyl radical 2. The C—C bond cleavage of iminyl radical 2 would provide cyanoalkyl radical 3, which reacted with SOMOphile giving rise to the desired products 128.

|
Download:
|
| Scheme 28. Photoinduced C—F/Cl bond formation with cycloketone oximes. | |
{kind=link}
6. C—B bond formation
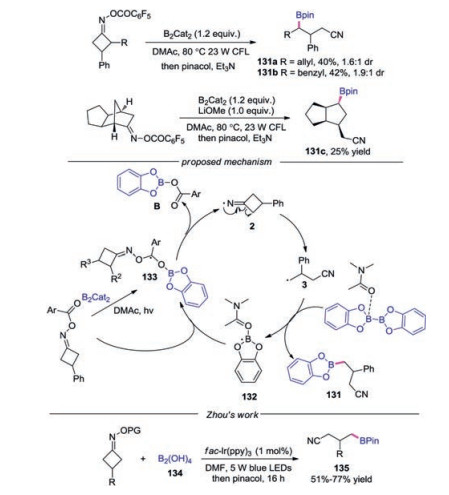
In 2019, Guo's group reported the transition-metalfree C—C bond scission/borylation of cyclobutanone oxime esters (Scheme 29) [54]. This method is amenable for the synthesis of both primary and secondary boronic esters. For the preparation of secondary boronic esters, light irradiation was essential to promote the reaction. A plausible radical chain propagation mechanism was proposed. Initially, radical intermediate 133 was formed through light-irradiation of B2(Cat)2·DMAc complex with cyclobutanone oxime ester. Then, iminyl radical 2 was generated by the cleavage of N—O bond, and the cyanoalkyl radical 3 was afforded through the C—C bond scission of iminyl radical 2. Subsequently, the radical 3 would react with B2(Cat)2·DMAc giving rise to the desired product 131 and DMAc-stabilized boryl radical 132. The resulting boryl radical 132 might propagate a radical chain process. The C—B bond formation with diboron was also reported by Zhou and co-workers [50].

|
Download:
|
| Scheme 29. Photoinduced C—B bond formation with cycloketone oximes. | |
{kind=link}
7. C—N bond formation
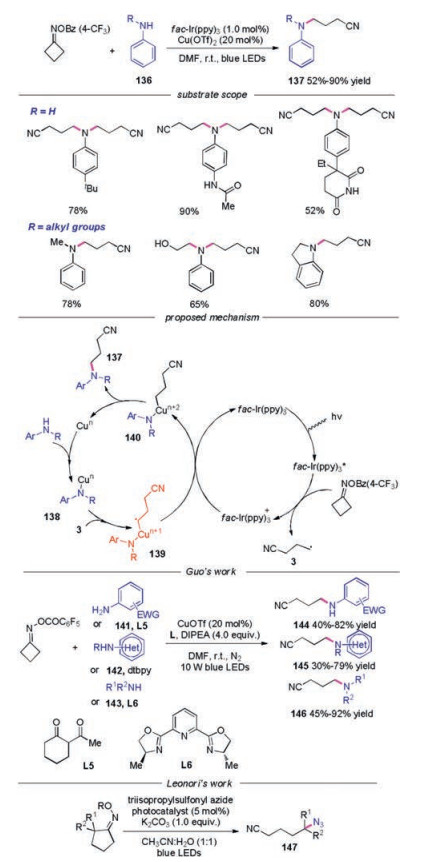
In 2017, Shi and co-workers developed a SET induced selective C(sp3)-N coupling with anilines via C-C bond cleavage under synergetic photoredox/copper-catalytic reaction systems (Scheme 30) [55]. A broad range of free anilines was examined, affording the dialkyl-functionalization products in good yields. Meanwhile, N-alkyl substituted anilines were workable in this reaction. Moreover, free alcohol and indoline were also suitable substrate under the conditions. A mechanism was proposed for this dual copper/photoredox catalytic cycle, as shown in Scheme 30. The distal radical 3 was generated by the reduction of photoexcited fac-Ir(ppy)3* followed by the C—C bond cleavage. Concurrently, the aniline coordinated with a low-valence copper catalyst to form a copper amido complex 138. Then, radical 3 would be trapped by copper amido complex 138 leading to intermediate 139. A highvalence copper alkylamido complex 140 would be obtained through a single-electron oxidation of complex 139 by photocatalyst, which would undergo reductive elimination to provide the desired product 137. In the same year, Guo's group reported the similar C—N bond formation between cycloketone oximes and aromatic amines or secondly amine under visible light conditions [56]. Leonori's group also described the C—N bond formation reaction of cycloketone oximes with azide compounds [53].

|
Download:
|
| Scheme 30. Photoinduced C—N bond formation with cycloketone oximes. | |
{kind=link}
8. Conclusion
In conclusion, recent advances for the synthesis of nitriles through photoinduced C—C bond cleavage of cycloketone oximes classified by the type of C—X bond forming are summarized. Various compounds possessing nitriles can be efficiently accessed via this method. The photoinduced conditions for the C—C bond cleavage of cycloketone oximes are mild, and the cyanoalkyl radical generated by single-electron-transfer and C—C bond cleavage is served as the key intermediate. A wide range of reactions with radical acceptors can be compatible under the conditions.
Despite the significant achievements in this field, there are still many issues to be solved. For example, the α-substituted carbon of cycloketone oximes was prochiral carbon after C—C bond cleavage, so is it possible to use this property to achieve asymmetric transformations? Furthermore, there is still room to investigate the asymmetric transformation of cyanoalkyl radical with in situ generated chiral compounds in catalytic way. Moreover, transformations on the alkyl chain of distal cyano group have yet been achieved, is there any useful transformation which utilize the carbon chain of distal cyano group? We wish this review would be useful for chemists who are interested in the synthetic transformation of cycloketone oximes.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21672037 and 21532001) and the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005) is gratefully acknowledged.
| [1] |
F.F. Fleming, Nat. Prod. Rep. 16 (1999) 597-606. DOI:10.1039/a804370a |
| [2] |
F.F. Fleming, L. Yao, P.C. Ravikumar, L. Funk, B.C. Shook, J. Med. Chem. 53 (2010) 7902-7917. DOI:10.1021/jm100762r |
| [3] |
R. López, C. Palomo, Angew. Chem. Int. Ed. 54 (2015) 13170-13184. DOI:10.1002/anie.201502493 |
| [4] |
L. Yu, H. Li, X. Zhang, et al., Org. Lett. 16 (2014) 1346-1349. DOI:10.1021/ol500075h |
| [5] |
J. Liu, H.X. Zheng, C.Z. Yao, B.F. Sun, Y.B. Kang, J. Am. Chem. Soc. 138 (2016) 3294-3297. DOI:10.1021/jacs.6b00180 |
| [6] |
Y.J. Zhuang, J. Liu, Y.B. Kang, Tetrahedron Lett. 57 (2016) 5700-5702. DOI:10.1016/j.tetlet.2016.11.034 |
| [7] |
J.J. Ge, C.Z. Yao, M.M. Wang, et al., Org. Lett. 18 (2016) 228-231. DOI:10.1021/acs.orglett.5b03367 |
| [8] |
Z. Shu, Y. Ye, Y. Deng, Y. Zhang, J. Wang, Angew. Chem. Int. Ed. 52 (2013) 10573-10576. DOI:10.1002/anie.201305731 |
| [9] |
S. Zhou, D. Addis, S. Das, K. Junge, M. Beller, Chem. Commum. (2009) 4883-4885. |
| [10] |
K.N.T. Tseng, A.M. Rizzi, N.K. Szymczak, J. Am. Chem. Soc. 135 (2013) 16352-16355. DOI:10.1021/ja409223a |
| [11] |
S. Guo, G. Wan, S. Sun, et al., Chem. Commum. 51 (2015) 5085-5088. DOI:10.1039/C5CC01024A |
| [12] |
W. Zhou, L. Zhang, N. Jiao, Angew. Chem. Int. Ed. 48 (2009) 7094-7097. DOI:10.1002/anie.200903838 |
| [13] |
L.V.A. Hale, T. Malakar, K.N.T. Tseng, et al., ACS Catal. 6 (2016) 4799-4813. DOI:10.1021/acscatal.6b01465 |
| [14] |
J. Boivin, E. Fouquet, S.Z. Zard, J. Am. Chem. Soc. 113 (1991) 1055-1057. DOI:10.1021/ja00003a057 |
| [15] |
W. Yin, X. Wang, New J. Chem. 43 (2019) 3254-3264. DOI:10.1039/C8NJ06165C |
| [16] |
X.Y. Yu, Q.Q. Zhao, J. Chen, W.J. Xiao, J.R. Chen, Acc. Chem. Res. 53 (2020) 1066-1083. DOI:10.1021/acs.accounts.0c00090 |
| [17] |
X.Y. Yu, J.R. Chen, W.J. Xiao, Chem. Rev. (2020), doi: http://dx.doi.org/10.1021/acs.chemrev.0c00030.
|
| [18] |
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [19] |
N.A. Romero, D.A. Nicewicz, Chem. Rev. 116 (2016) 10075-10166. DOI:10.1021/acs.chemrev.6b00057 |
| [20] |
W. Yang, B. Li, M. Zhang, S. Wang, S. Yuan, Chin. Chem. Lett. 31 (2020) 1313-1316. DOI:10.1016/j.cclet.2019.10.022 |
| [21] |
L. Li, H. Chen, M. Mei, L. Zhou, Chem. Commun. 53 (2017) 11544-11547. DOI:10.1039/C7CC07347J |
| [22] |
B.Q. He, X.Y. Yu, P.Z. Wang, J.R. Chen, W.J. Xiao, Chem. Commun. 54 (2018) 12262-12265. DOI:10.1039/C8CC07072E |
| [23] |
B. Zhao, Z. Shi, Angew. Chem. Int. Ed. 56 (2017) 12727-12733. DOI:10.1002/anie.201707181 |
| [24] |
X.Y. Yu, J.R. Chen, P.Z. Wang, et al., Angew. Chem. Int. Ed. 57 (2018) 738-743. DOI:10.1002/anie.201710618 |
| [25] |
P.Z. Wang, X.Y. Yu, C.Y. Li, et al., Chem. Commun. 54 (2018) 9925-9928. DOI:10.1039/C8CC06145A |
| [26] |
S. Yao, K. Zhang, Q.Q. Zhou, et al., Chem. Commun. 54 (2018) 8096-8099. DOI:10.1039/C8CC04503H |
| [27] |
X.Y. Yu, Q.Q. Zhao, J. Chen, J.R. Chen, W.J. Xiao, Angew. Chem. Int. Ed. 57 (2018) 15505-15509. DOI:10.1002/anie.201809820 |
| [28] |
J. Chen, B.Q. He, P.Z. Wang, et al., Org. Lett. 21 (2019) 4359-4364. DOI:10.1021/acs.orglett.9b01529 |
| [29] |
X. Shen, J.J. Zhao, S. Yu, Org. Lett. 20 (2018) 5523-5527. DOI:10.1021/acs.orglett.8b02540 |
| [30] |
W.L. Xing, R. Shang, G.Z. Wang, Y. Fu, Chem. Commun. 55 (2019) 14291-14294. DOI:10.1039/C9CC08077E |
| [31] |
M.M. Zhang, S.H. Li, J.L. Tu, Q.Q. Min, F. Liu, Org. Chem. Front. 7 (2020) 622-627. DOI:10.1039/C9QO01446B |
| [32] |
Y. Jian, M. Chen, C. Yang, W.J. Xia, Eur. J. Org. Chem. 2020 (2020) 1439-1442. DOI:10.1002/ejoc.201900406 |
| [33] |
W. Zhang, Y.L. Pan, C. Yang, X. Li, B. Wang, Org. Chem. Front. 6 (2019) 2765-2770. DOI:10.1039/C9QO00625G |
| [34] |
B. Zhao, X. Kong, B. Xu, Tetrahedron Lett. 60 (2019) 2063-2066. DOI:10.1016/j.tetlet.2019.06.059 |
| [35] |
T. Wang, Y.N. Wang, R. Wang, et al., Nat. Commun. 10 (2019) 5373-5381. DOI:10.1038/s41467-019-13369-x |
| [36] |
B. Lu, Y. Cheng, L.Y. Chen, J.R. Chen, W.J. Xiao, ACS Catal. 9 (2019) 8159-8164. DOI:10.1021/acscatal.9b02830 |
| [37] |
B. Zhao, Y. Wu, Y. Yuan, Z. Shi, Chem. Commun. 56 (2020) 4676-4679. DOI:10.1039/D0CC00988A |
| [38] |
F. Le Vaillant, M. Garreau, S. Nicolai, et al., Chem. Sci. 9 (2018) 5883-5889. DOI:10.1039/C8SC01818A |
| [39] |
Y. He, D. Anand, Z. Sun, L. Zhou, Y. He, Org. Lett. 21 (2019) 3769-3773. DOI:10.1021/acs.orglett.9b01210 |
| [40] |
E.M. Dauncey, S.U. Dighe, J.J. Douglas, D. Leonori, Chem. Sci. 10 (2019) 7728-7733. DOI:10.1039/C9SC02616A |
| [41] |
Y. Wang, J. Shen, Q. Chen, L. Wang, M. He, Chin. Chem. Lett. 30 (2019) 409-412. DOI:10.1016/j.cclet.2018.09.009 |
| [42] |
P.Z. Wang, B.Q. He, Y. Cheng, J.R. Chen, W.J. Xiao, Org. Lett. 21 (2019) 6924-6929. DOI:10.1021/acs.orglett.9b02535 |
| [43] |
P.J. Xia, Z.P. Ye, Y.Z. Hu, et al., Org. Lett. 21 (2019) 2658-2662. DOI:10.1021/acs.orglett.9b00651 |
| [44] |
X.Y. Yu, P.Z. Wang, D.M. Yan, et al., Adv. Synth. Catal. 360 (2018) 3601-3606. DOI:10.1002/adsc.201800834 |
| [45] |
W. Zhang, C. Yang, Z.P. Zhang, X. Li, J.P. Cheng, Org. Lett. 21 (2019) 4137-4142. DOI:10.1021/acs.orglett.9b01325 |
| [46] |
X.-M. Xu, D.-M. Chen, Z.-L. Wang, Chin. Chem. Lett. 31 (2020) 49-57. DOI:10.1016/j.cclet.2019.05.048 |
| [47] |
J. Zhang, X. Li, W. Xie, S. Ye, J. Wu, Org. Lett. 21 (2019) 4950-4954. DOI:10.1021/acs.orglett.9b01323 |
| [48] |
Y. Liu, Q.L. Wang, Z. Chen, et al., Chem. Commun. 56 (2020) 3011-3014. DOI:10.1039/C9CC10057A |
| [49] |
M. Zheng, G. Li, H. Lu, Org. Lett. 21 (2019) 1216-1220. DOI:10.1021/acs.orglett.9b00201 |
| [50] |
D. Anand, Y. He, L. Li, L. Zhou, Org. Biomol. Chem. 17 (2019) 533-540. DOI:10.1039/C8OB02987C |
| [51] |
B. Zhao, C. Chen, J. Lv, et al., Org. Chem. Front. 5 (2018) 2719-2722. DOI:10.1039/C8QO00747K |
| [52] |
B. Zhao, H. Tan, C. Chen, N. Jiao, Z. Shi, Chin. J. Chem. 36 (2018) 995-999. DOI:10.1002/cjoc.201800206 |
| [53] |
E.M. Dauncey, S.P. Morcillo, J.J. Douglas, N.S. Sheikh, D. Leonori, Angew. Chem. Int. Ed. 57 (2018) 744-748. DOI:10.1002/anie.201710790 |
| [54] |
J.J. Zhang, X.H. Duan, Y. Wu, J.C. Yang, L.N. Guo, Chem. Sci. 10 (2019) 161-166. DOI:10.1039/C8SC03315C |
| [55] |
B. Zhao, M. Wang, Z. Shi, J. Org. Chem. 84 (2019) 10145-10159. DOI:10.1021/acs.joc.9b01338 |
| [56] |
L. Yang, J.Y. Zhang, X.H. Duan, et al., J. Org. Chem. 84 (2019) 8615-8629. DOI:10.1021/acs.joc.9b01084 |