 2020, Vol. 31
2020, Vol. 31 


b College of Chemistry, Xiangtan University, Xiangtan 411105, China;
c State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
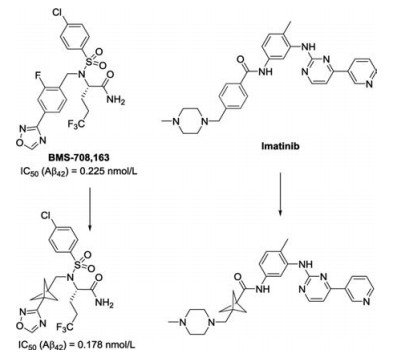
Installation of three-dimensional carbon framework into the structure of drug candidates has attracted increasing attention in drug discovery, thus altering the pharmacokinetic properties [1-4]. As the smallest member of propellane family, the highly strained [1.1.1]propellane 1 possess unique reactivity across the central bond. To perfect ring opening processes of [1.1.1]propellane 1, tremendous efforts have been devoted to the development of efficient synthetic methodologies leading to highly functionalized bicyclo[1.1.1]pentane (BCP) and cyclobutane derivatives [5-8]. Among them, 1, 3-disubstituted bicyclo[1.1.1]pentane motif has emerged as bioisosteres for phenyl, tert-butyl and alkyne functional groups in modern medicinal chemistry. For example, the replacement of a para-substituted fluorophenyl ring in the γ-secretase inhibitor BMS708, 163 with a bicyclo[1.1.1]pentane moiety results in improved aqueous solubility and passive permeability, as well as improved oral absorption in a mouse model of γ-secretase inhibition [9]. The bicyclo[1.1.1]pentane analogue of the anti-leukemia drug imatinib was found to exhibit higher themodynamic solubility (Scheme 1) [10].

|
Download:
|
| Scheme 1. Bicyclo[1.1.1]pentane as a bioisostere for a phenyl ring in medicinal chemistry. | |
{kind=link}
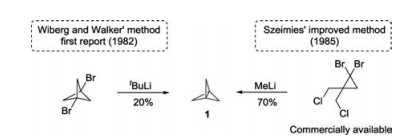
The first synthesis of [1.1.1]propellane 1 was reported by Wiberg and Walker in 1982 via reductive intramolecular coupling of 1, 3-dibromobicyclo[1.1.1]pentane [11]. Later in 1985, Szeimies and coworkers developed an improved method for the preparation of [1.1.1]propellane 1 by using the commercially available 1, 1-dibromo-2, 2-bis(chloromethyl)cyclopropane as the starting material (Scheme 2) [12, 13].

|
Download:
|
| Scheme 2. The reported routes for the synthesis of [1.1.1]propellane. | |
{kind=link}
In recent years, the synthetic and medicinal community have witnessed fast and impressive development of the [1.1.1]propellane chemistry, and a variety of approaches for the construction of bicyclo[1.1.1]pentane and cyclobutane derivatives have been realized. Although several reviews focusing on the synthesis of bicyclo[1.1.1]pentane derivatives have recently been reported [6, 7], the applications of [1.1.1]propellane 1 in organic synthesis still lacks a concise review in this fast growing field. This review herein is aimed to summarize recent progress in the field of synthetic transformations starting from [1.1.1]propellane 1, highlighting three different reaction pathways for the cleavage of the central bond.
2. Radical addition to the central bond of [1.1.1]propellaneAs mentioned above, radical addition to [1.1.1]propellane 1 represents the most frequently observed reaction attributed to its high reactivity, with many different radicals and radical sources being reported including halides, chalcogenides to carboncentered species. In this transformation, the central bond of [1.1.1]propellane 1 is broken to yield kinetically stable bicyclo [1.1.1]-pentyl radical, giving rise to various functionalized bicyclo [1.1.1]-pentane derivatives. It is noteworthy that the oligomerization side products could be observed when the low chain-transfer constant of free-radical addition pathway occurred. In 2017, Uchiyama and co-workers developed a radical multicomponent carboamination of [1.1.1]propellane 1 by employing iron(Ⅱ) phthalocyanine as the catalyst and TBHP as the oxidant (Scheme 3) [14]. Under the mild conditions, hydrazyl reagent 2 was used as a radical precursor with di-tert-butyl azodicarboxylate 3 as a radical acceptor for the construction of multi-functionalized bicyclo[1.1.1] pentane derivatives 4, which could be further transformed into synthetically useful 3-substituted BCP-amines. On the basis of the experimental observations and DFT calculations, hydrazines were shown to efficiently undergo oxidative denitrogenation under iron (Ⅱ) phthalocyanine and TBHP to generate the free radical 5 in situ. Radical addition of 5 with [1.1.1]propellane 1 gave rise to the BCP-radical intermediate 6, which could be trapped by di-tert-butyl azodicarboxylate 3 to yield the desired product 4.

|
Download:
|
| Scheme 3. Iron(Ⅱ)-catalyzed multicomponent carboamination of [1.1.1]propellane 1. | |
{kind=link}
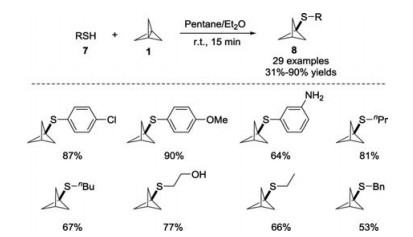
The reaction of [1.1.1]propellane 1 with thiophenol 7 was first achieved by Szeimies and co-workers in 1985, providing an effective tool for the quantification of propellane solutions [12]. In 2018, the group of Bräse further investigated the reaction in details and enlarged the scope of thiols 7, which showed that halo-, hydroxy-, methoxy-, carboxy-, amino- and nitro-substituted thiols were well tolerated (Scheme 4) [15]. The reaction proceeded at room temperature without any catalyst, and no by-products were detected. The deuterium labelling experiments revealed that the reaction underwent a radical chain process.

|
Download:
|
| Scheme 4. Addition of thiols with [1.1.1]propellane 1 to yield BCP sulfides. | |
{kind=link}
Recently, the synthetic utility of this method was further demonstrated through the conversion of BCP sulfides 8 into the corresponding sulfoximines 9 (Scheme 5) [16]. Moreover, copper(Ⅰ)-catalyzed N-arylations of BCP sulfoximines 9 with aryl halides were carried out under optimized conditions, showing a wide substrate scope.

|
Download:
|
| Scheme 5. Formation of BCP sulfoximines applied in copper(Ⅰ)-catalyzed N-arylation. | |
{kind=link}
Additionally, the same group reported the UV-initiated radical reaction for the synthesis of symmetrical and unsymmetrical 1, 3-bissulfanylbicyclo[1.1.1]pentanes from disulfides 11 and [1.1.1]propellane 1 (Scheme 6) [17]. By changing the ratio of [1.1.1]propellane to disulfide, the reaction could provide only BCP product or a mixture of BCPs and [2]staffanes. However, the separation of these products was challenging due to the similar polarity. The authors also proposed a possible reaction mechanism to explain the formation of different products.

|
Download:
|
| Scheme 6. UV-promoted radical reaction of disulfides and [1.1.1]propellane 1. | |
{kind=link}
In 2018, Anderson and co-workers described an efficient triethylborane-initiated atom-transfer radical addition of [1.1.1]propellane 1 with alkyl halides 15 to access 1-halo-3-substituted bicyclo[1.1.1]pentanes 16 (Scheme 7) [18]. This protocol exhibited a broad substrate scope and good functional group tolerance. Furthermore, the synthetic applicability of this process was also demonstrated through late-functionalization of complex molecules such as nucleosides, peptides and drug-like molecules.

|
Download:
|
| Scheme 7. Synthesis of 1-halo-3-substituted bicyclo[1.1.1]pentanes through triethylborane-initiated radical addition pathway. | |
{kind=link}
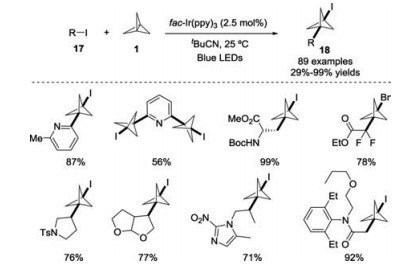
Subsequently, the same group further disclosed a visible light promoted radical addition of organic halides 17 with [1.1.1]propellane 1 for the functionalization of carbon-carbon σ-bonds (Scheme 8) [19]. Under mild conditions, both (hetero)aryl radicals and nonstabilized alkyl radicals were generated in situ by using fac-Ir(ppy)3 as the photocatalyst, affording a variety of (hetero) arylated and polycyclic BCP products 18 with abroad substrate scope. Additionally, the importance of the photocatalyst in this process was demonstrated according to the mechanistic investigations.

|
Download:
|
| Scheme 8. Photoredox-catalyzed radical reaction of organic halides with [1.1.1]propellane 1. | |
{kind=link}
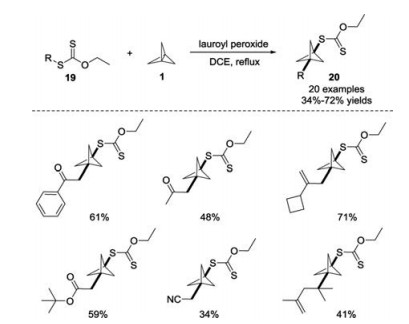
In 2019, Riant and co-workers reported a radical exchange strategy by incorporating xanthate derivatives 19 into [1.1.1]propellane 1 under metal-free conditions (Scheme 9) [20]. By employing substoichiometric amounts of dilauroyl peroxide (DLP) as a radical initiator, this method enabled the synthesis of functionalized BCP derivatives with xanthate moieties. The reaction featured a broad substrate scope and functional group tolerance respect to the xanthate moiety.

|
Download:
|
| Scheme 9. Metal-free synthesis of BCP xanthate derivatives in the presence of dilauroyl peroxide. | |
{kind=link}
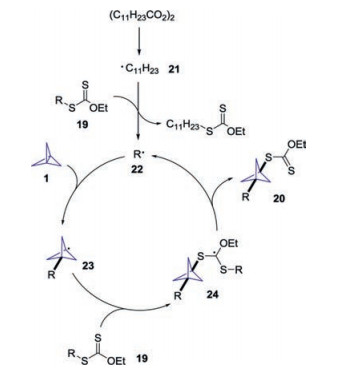
On the basis of mechanistic experiments, the authors proposed a plausible mechanism for this transformation (Scheme 10). Initially, DLP would undergo fragmentation/decarboxylation to provide the decyl radical 21, which would react with xanthate 19 to generate radical 22 in situ. The addition of radical 22 with [1.1.1]propellane 1 would give rise to the radical intermediate 23. Subsequently, the reaction of radical 23 with xanthate 19 would produce the stabilized intermediate radical 24, followed by radical cleavage to produce the BCP product 20.

|
Download:
|
| Scheme 10. Proposed mechanism for the formation of BCP xanthate derivatives. | |
{kind=link}
The UV-initiated silaboration of [1.1.1]propellane 1 to access BCP derivatives bearing B and Si functional groups was achieved by Uchiyama and co-workers (Scheme 11) [21]. Under mild and additive-free conditions, the unsymmetrically silaborated products 26 could be obtained on a gram-scale without the need for columnchromatographic purification. On the basis of experimental studies and DFT calculations, it was suggested that a radical chain mechanism was involved in this transformation. Notably, the synthetic applicability of the process was also demonstrated through the conversion of the C–B/C–Si bonds on the BCP scaffold into various functional groups. In particular, by combination of highly activated BCP boronic esters, Cu2O, and PdCl2(dppf) catalyst, the authors developed a facile approach for Suzuki–Miyaura crosscoupling reaction at the highly sterically hindered BCP bridgehead sp3 carbon center.

|
Download:
|
| Scheme 11. UV-initiated silaboration of [1.1.1]propellane 1 and further applications. | |
{kind=link}
Bicyclo[1.1.1]pentylamine has been considered as a high-value bioisostere for aniline to improve antimetabolic clearance activity in modern medicinal chemistry. Recently, Leonori and co-workers reported the first example of visible-light-promoted radical strain-release-functionalization strategyfor the divergent construction of 1, 3-disubstituted bicyclo[1.1.1]pentylamines 28 (Scheme 12) [22]. The reaction tolerated a variety of N-substituted amides 27, which served as precursors for generating amidyl radicals, representing an umpolung approach to ionic strain-release amination. Accordingly, the putative mechanism revealed that the visible-light excited photocatalyst was quenched by radical precursor 29 via a single electron transfer (SET) to provide the amidyl radical 30. The addition of amidyl radical 30 to [1.1.1]propellane 1 would produce stabilized radical intermediate 31, followed by atom/group-transfer reaction (SH2) with SOMOphiles (X–Y) to deliver the desired product 28. Finally, the electron poor radical Y· would undergo reduction by the reduced state of photocatalyst to complete the catalytic cycle.

|
Download:
|
| Scheme 12. Visible-light-mediated strain-release multicomponent reaction of [1.1.1]propellane 1 with electrophilic nitrogen-radicals. | |
{kind=link}
Most recently, the MacMillan group developed a three-component radical coupling of [1.1.1]propellane 1 in the presence of dual photoredox/copper catalysis (Scheme 13) [23]. This elegant protocol enabled the one-step synthesis of diverse functionalized BCP derivatives 32 within short timescales (5 min to 1 h). Remarkably, the reaction featured a broad substrate scope and good functional group tolerance, and a variety of radical precursors including those which generated alkyl, α-acyl, trifloromethyl, and sulfonyl radicals as well as multiple classes of N-, P-, and S-nucleophiles were proven to amenable for this transformation. Moreover, this metallaphotoredox catalysis strategy was successfully applied in the late-stage functionalization of drugs and natural products and preparation of pharmaceutical analogues.

|
Download:
|
| Scheme 13. Dual photoredox/copper catalyzed three-component radical coupling of [1.1.1]propellane 1. | |
{kind=link}
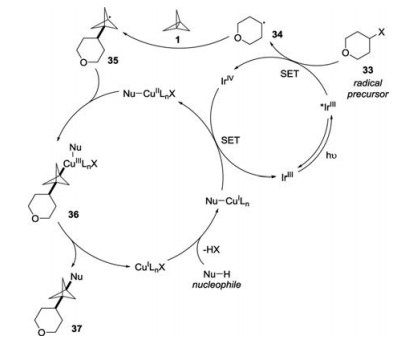
The mechanism for this multicomponent transformation was proposed as follows: firstly, the alkyl radical 34 was generated through the SET reduction of iodoniumdicarboxylate 33 (Epc [33/33*-] = -0.82 V vs. SCE in acetonitrile) by excited *IrⅢ (E1/2red [IrⅣ/*IrⅢ] = -1.81 V vs. SCE in acetonitrile). Subsequently, the addition of alkyl radical 34 to [1.1.1]propellane 1 would afford stabilized radical intermediate 35. The combination of radical intermediate 35 with nucleophile-ligated copper complex would lead to CuⅢ species 36, which would go through reductive elimination to afford the desired product 37. Finally, the nucleophile-ligated CuI complex was oxidized by IrⅣ (E1/2red [IrⅣ/IrⅢ] = +0.77 V vs. SCE in acetonitrile) to provide CuⅡ complex with the release of IrⅢ species to facilitate both catalytic cycles (Scheme 14).

|
Download:
|
| Scheme 14. Proposed mechanism for the synthesis of polysubstituted BCP products. | |
{kind=link}
3. Nucleophilic addition to the central bond of [1.1.1]propellane
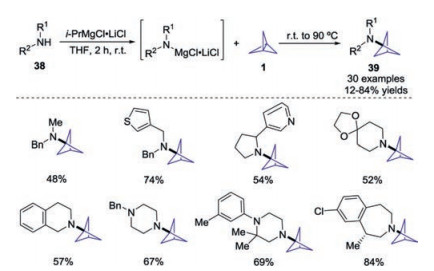
Considering the importance of BCP functionality, nucleophilic addition to the central bond of [1.1.1]propellane 1 has become an alternative and complementary method for that of radical additions. The breakthrough was made by Baran and co-workers in 2016, and they found that [1.1.1]propellane 1 would undergo ionic strain-release amination by deprotonated dialkyl amines, solving a long standing issue for the synthesis of monosubstituted bicyclo[1.1.1]pentylamines 39 (Scheme 15) [24]. It was found that the use of turbo-amides was the key to complete the transformation.

|
Download:
|
| Scheme 15. Turbo-amide enabled ring opening of [1.1.1]propellane 1 to access mono-substituted bicyclo[1.1.1]pentylamines. | |
{kind=link}
The aryl-substituted BCPs 40 could be prepared according to Baran's method using aryl Grignard reagent instead of turboamides. Recently, the synthetic utility of aryl-substituted BCPs 40 was demonstrated by Davies and co-workers, through enantioselective intermolecular C(sp3)–H insertion reactions of donor/acceptor diazo compounds 41 (Scheme 16) [25]. By employing the chiral dirhodium complex Rh2(TCPTAD)4 as the catalyst, a variety of 1, 3-disubstituted BCPs 42 bearing a proximal stereocenter were obtained with up to 99% yield and 94% ee.

|
Download:
|
| Scheme 16. Chiral dirhodium complex-catalyzed asymmetric C–H functionalization of BCPs towards chiral substituted BCPs. | |
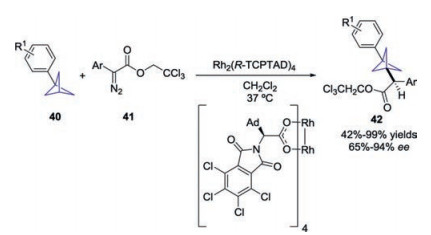{kind=link}
In 2017, Knochel and co-workers developed an elegant and practical method for the synthesis of diarylated BCP derivatives 44 through Grignard reagent-mediated ring-opening of [1.1.1]propellane 1 and Negishi coupling after transmetallation with ZnCl2 (Scheme 17) [26]. The bis-arylated BCPs could be regarded as potential bioisosteres of internal alkynes. For instance, BCP analogues of tazarotene and the metabotropic glutamate receptor 5 (mGluR5) antagonist were prepared and their physicochemical properties were evaluated.

|
Download:
|
| Scheme 17. Synthesis of diarylated BCP derivatives. | |
{kind=link}
In 2018, the Walsh group developed a new method for the synthesis of BCP benzylamine derivatives 46 through a reaction of [1.1.1]propellane 1 with 2-azaallyl anions generated in situ from N-benzyl ketimines 45 (Scheme 18) [27]. The reaction proceeded rapidly with high yields and tolerated a broad substrate scope at room temperature. On the basis of their previous work, the authorsproposed several possible mechanisms for this transformation. In one possible pathway, 2-azaallyl anion 47 generated from deprotonation of N-benzyl ketimine 45 would undergo a single electron transfer (SET) with [1.1.1]propellane to form azaallyl radical 48 and [1.1.1]propellane radical anion. Subsequently, radical-radical coupling of both species would produce the anion intermediate 49, followed by protonation at the bridgehead carbon to furnish the desired product 46 and regenerate 2-azaallyl anion 47.

|
Download:
|
| Scheme 18. Additions of 2-azaallyl anions with [1.1.1]propellane 1 to synthesize BCP benzylamines. | |
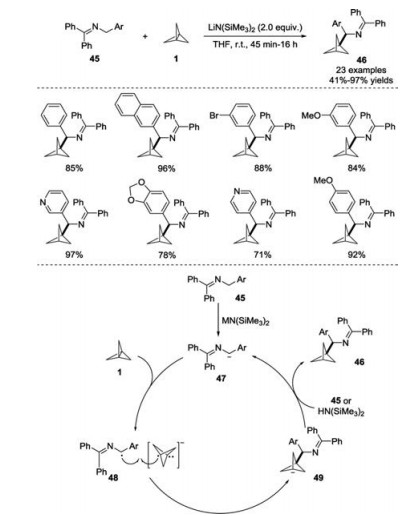{kind=link}
Later, the same group extended this strategy to the preparation of BCP-containing dithianes 51 (Scheme 19) [28]. The reaction of 2-aryl-1, 3-dithianes 50 and [1.1.1]propellane 1 featured a broad substrate scope and was amenable to scale up. The utility of the products was further demonstrated by the conversion of the dithiane moieties into a variety of functional groups. Additionally, on the basis of computational results, it was proposed that a two-electron pathway might be involved in this transformation.

|
Download:
|
| Scheme 19. Reactions of 2-aryl-1, 3-dithianes and [1.1.1]propellane 1 to synthesize BCP dithianes. | |
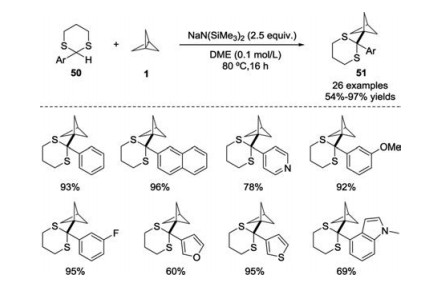{kind=link}
In 2019, Gleason and co-workers disclosed a one-pot aminoalkylation of [1.1.1]propellane 1 toward the synthesis of 3-alkylbicyclo[1.1.1]pentan-1-amines 53 under mild reaction conditions (Scheme 20) [29]. This protocol exhibited a broad substrate scope, providing a facile approach for accessing 1, 3-difunctionalized BCPs containing high-value functional groups.

|
Download:
|
| Scheme 20. Aminoalkylation of [1.1.1]propellane 1 for the synthesis of 3-alkylbicyclo[1.1.1]pentan-1-amines. | |
{kind=link}
Earlier this year, Aggarwal and co-workers reported a transition metal-free three-component of [1.1.1]propellane 1 with organometallic reagents and organoboronic esters for the rapid synthesis of 1, 3-disubstituted BCP derivatives 55 (Scheme 21) [30]. During the reaction process, boronate complexes 54 were generated in situ from the BCP-metal species and boronic esters, which would undergo electrophile-induced 1, 2-metallate rearrangement for the construction of C—C bonds. Furthermore, this strategy could be extended to access tertiary boronic ester-substituted BCPs containing contiguous quaternary centers via a four-component coupling of alkenyl BCP-boronate complexes with a variety of electrophiles.

|
Download:
|
| Scheme 21. Synthesis of 1, 3-disubstituted BCPs through 1, 2-metallate rearrangements of boronate complexes. | |
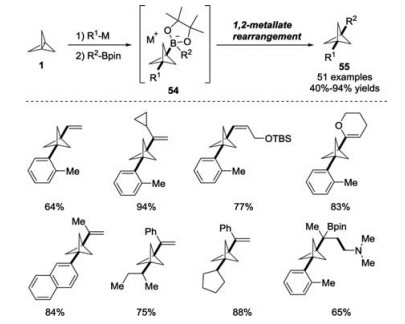{kind=link}
4. Transition metal-catalyzed transformations of [1.1.1]propellane
Contrary to the well-developed methods for the preparation of functionalized BCP derivatives through radical or carbanion pathways, transition metal-catalyzed transformation of [1.1.1]propellane are less established. In 2019, Aggarwal, Bedford and co-workers described a nickel-catalyzed cyclopropanation of alkenes 56 with [1.1.1]propellane 1 as a carbene precursor (Scheme 22) [31]. The reaction exhibited a broad functional group tolerance under mild conditions, leading to the formation of methylenespiro[2.3]hexane derivatives 57. Both experimental and computational results supported the formation of a nickel carbene through a concerted double C—C bond cleavage of a nickel-[1.1.1] propellane complex.

|
Download:
|
| Scheme 22. Nickel-catalyzed cyclopropanation of alkenes with [1.1.1]propellane 1. | |
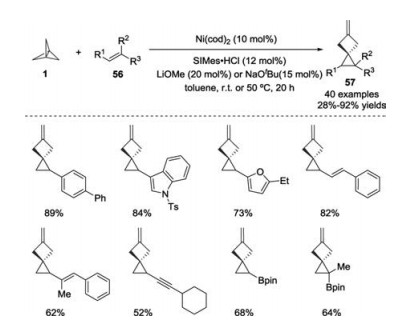{kind=link}
Almost the same time, a copper-catalyzed formation of cyclobutane-containing alkynes 59 and allenes 60 from the ring opening reactions of [1.1.1]propellane 1 with alkynes 58 was disclosed by Tolnai and co-workers (Scheme 23) [32].

|
Download:
|
| Scheme 23. Copper-catalyzed divergent synthesis of cyclobutane-containing alkynes and allenes. | |
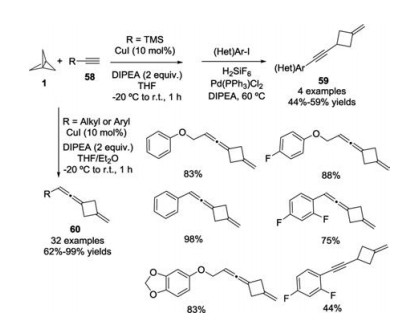{kind=link}
5. Conclusion
In summary, this review highlights the most recent advances of the synthetic applications of [1.1.1]propellane. Overall, a number of different approaches for the synthesis of BCP and cyclobutane derivatives from [1.1.1]propellane have been conceived and developed. Generally, the recent progress of this chemistry shows that the weak central bond of [1.1.1]propellane can be broken either through a radical, anionic or transition metal-catalyzed pathway. For example, radical addition to [1.1.1]propellane produces stabilized BCP-radical intermediate, which can be subsequently trapped by a radical acceptor or transition metal catalyst. Because of its inherent structural features and unique reactivity profile, [1.1.1]propellane possesses a very important role in both synthetic and medicinal community for incorporating a three-dimensional cyclic scaffold into small molecules as bioisosteres.
Despite the recent achievements in this field, there are still many issues existed, which are anticipated to be solved for the chemistry of [1.1.1]propellane. For instance, the synthesis of optically pure compounds bearing a BCP moiety through one-pot multicomponent catalytic asymmetric transformation of [1.1.1]propellane has yet not to be achieved. Furthermore, the development of sustainable processes based on flow chemistry or electrochemistry as well as the discovery of new reactivities with [1.1.1]propellane can be expected in the near future.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (Nos. 21672037 and 21532001) and the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (No. 2019R01005) is gratefully acknowledged.
| [1] |
M.R. Barbachyn, D.K. Hutchinson, D.S. Toops, et al., Bioorg. Med. Chem. Lett. 3 (1993) 671-676. DOI:10.1016/S0960-894X(01)81251-8 |
| [2] |
Y.L. Goh, Y.T. Cui, V. Pendharkar, V.A. Adsool, ACS Med. Chem. Lett. 8 (2017) 516-520. DOI:10.1021/acsmedchemlett.7b00018 |
| [3] |
N.D. Measom, K.D. Down, D.J. Hirst, et al., ACS Med. Chem. Lett. 8 (2017) 43-48. DOI:10.1021/acsmedchemlett.6b00281 |
| [4] |
S.O. Kokhan, A.V. Tymtsunik, S.L. Grage, et al., Angew. Chem. Int. Ed. 55 (2016) 14788-14792. DOI:10.1002/anie.201608116 |
| [5] |
A.M. Dilmaç, E. Spuling, A. de Meijere, S. Bräse, Angew. Chem. Int. Ed. 56 (2017) 5684-5718. DOI:10.1002/anie.201603951 |
| [6] |
J. Kanazawa, M. Uchiyama, Synlett 30 (2019) 1-11. DOI:10.1055/s-0037-1610314 |
| [7] |
X. Ma, L. Nhat Pham, Asian J. Org. Chem. 9 (2020) 8-22. DOI:10.1002/ajoc.201900589 |
| [8] |
P.K. Mykhailiuk, Org. Biomol. Chem. 17 (2019) 2839-2849. DOI:10.1039/C8OB02812E |
| [9] |
A.F. Stepan, C. Subramanyam, I.V. Efremov, et al., J. Med. Chem. 55 (2012) 3414-3424. DOI:10.1021/jm300094u |
| [10] |
K.C. Nicolaou, D. Vourloumis, S. Totokotsopoulos, et al., ChemMedChem 11 (2016) 31-37. DOI:10.1002/cmdc.201500510 |
| [11] |
K.B. Wiberg, F.H. Walker, J. Am. Chem. Soc. 104 (1982) 5239-5240. DOI:10.1021/ja00383a046 |
| [12] |
K. Semmler, G. Szeimies, J. Belzner, J. Am. Chem. Soc. 107 (1985) 6410-6411. DOI:10.1021/ja00308a053 |
| [13] |
J. Belzner, U. Bunz, K. Semmler, et al., Chem. Ber. 122 (1989) 397-398. DOI:10.1002/cber.19891220233 |
| [14] |
J. Kanazawa, K. Maeda, M. Uchiyama, J.Am.Chem. Soc. 139 (2017) 17791-17794. DOI:10.1021/jacs.7b11865 |
| [15] |
R.M. Bär, S. Kirschner, M. Nieger, S. Bräse, Chem. Eur. J. 24 (2018) 1373-1382. DOI:10.1002/chem.201704105 |
| [16] |
(a) R.M. Bär, L. Langer, M. Nieger, S. Bräse, Adv. Synth. Catal. (2020), doi: http://dx.doi.org/10.1002/adsc.201901453; (b) R.M. Bär, P.J. Gross, M. Nieger, S. Bräse, Chem. Eur. J. 26 (2020) 4242-4245. |
| [17] |
R.M. Bar, G. Heinrich, M. Nieger, O. Fuhr, S. Brase, Beilstein J. Org. Chem. 15 (2019) 1172-1180. DOI:10.3762/bjoc.15.114 |
| [18] |
D.F.J. Caputo, C. Arroniz, A.B. Dürr, et al., Chem. Sci. 9 (2018) 5295-5300. DOI:10.1039/C8SC01355A |
| [19] |
J. Nugent, C. Arroniz, B.R. Shire, et al., ACS Catal. 9 (2019) 9568-9574. DOI:10.1021/acscatal.9b03190 |
| [20] |
S.K. Rout, G. Marghem, J. Lan, T. Leyssens, O. Riant, Chem. Commun. (Camb.) 55 (2019) 14976-14979. DOI:10.1039/C9CC07610G |
| [21] |
M. Kondo, J. Kanazawa, T. Ichikawa, et al., Angew. Chem. Int. Ed. 59 (2020) 1970-1974. DOI:10.1002/anie.201909655 |
| [22] |
J.H. Kim, A. Ruffoni, Y. Al-Faiyz, N.S. Sheikh, D. Leonori, Angew. Chem. Int. Ed. 59 (2020) 8225-8231. DOI:10.1002/anie.202000140 |
| [23] |
X. Zhang, R.T. Smith, C. Le, et al., Nature 580 (2020) 220-226. DOI:10.1038/s41586-020-2060-z |
| [24] |
R. Gianatassio, J.M. Lopchuk, J. Wang, et al., Science 351 (2016) 241. DOI:10.1126/science.aad6252 |
| [25] |
Z.J. Garlets, J.N. Sanders, H. Malik, et al., Nat. Catal. 3 (2020) 351-357. DOI:10.1038/s41929-019-0417-1 |
| [26] |
I.S. Makarov, C.E. Brocklehurst, K. Karaghiosoff, G. Koch, P. Knochel, Angew. Chem. Int. Ed. 56 (2017) 12774-12777. DOI:10.1002/anie.201706799 |
| [27] |
R.A. Shelp, P.J. Walsh, Angew. Chem. Int. Ed. 57 (2018) 15857-15861. DOI:10.1002/anie.201810061 |
| [28] |
N. Trongsiriwat, Y. Pu, Y. Nieves-Quinones, et al., Angew. Chem. Int. Ed. 58 (2019) 13416-13420. DOI:10.1002/anie.201905531 |
| [29] |
J.M.E. Hughes, D.A. Scarlata, A.C.Y. Chen, J.D. Burch, J.L. Gleason, Org. Lett. 21 (2019) 6800-6804. DOI:10.1021/acs.orglett.9b02426 |
| [30] |
S. Yu, C. Jing, A. Noble, V.K. Aggarwal, Angew. Chem. Int. Ed. 59 (2020) 3917-3921. DOI:10.1002/anie.201914875 |
| [31] |
S. Yu, A. Noble, R.B. Bedford, V.K. Aggarwal, J. Am. Chem. Soc. 141 (2019) 20325-20334. DOI:10.1021/jacs.9b10689 |
| [32] |
D. Lasányi, G.L. Tolnai, Org. Lett. 21 (2019) 10057-10062. DOI:10.1021/acs.orglett.9b03999 |