 2020, Vol. 31
2020, Vol. 31 


b College of Medical Imaging, Shanxi Medical University, Taiyuan 030001, China;
c Institute of Medical Engineering, Department of Biophysics, School of Basic Medical Science, Health Science Center, Xi'an Jiaotong University, Xi'an 710061, China;
d College of Chemistry and Life Science, Institute of Functional Molecules, Chengdu Normal University, Chengdu 611130, China;
e Division of Pharmaceutics, National Pharmaceutical Engineering Center for Solid Preparation in Chinese Herbal Medicine, Jiangxi University of Traditional Chinese Medicine, Nanchang 330006, China
Cancer is one of the most serious diseases threatening human health globally, and the morbidity and mortality of this disease continue to rise each year [1]. Early diagnosis and effective treatment are essential for better health management. Currently, conventional chemotherapy is the mainstream treatment designed to target highly proliferating neoplastic cells [2, 3], but chemotherapy has many shortcomings that hinder its clinical translation, such as limited drug solubility, nonspecific distribution, weak bioavailability, rapid blood clearance and inevitable tissue toxicity [4, 5]. To address these issues, pharmacists have designed novel drug delivery systems (DDS) with the help of rapidly developing nanomedicine, to direct bioactive components to tumor sites [6-10]. Despite promising results, several serious obstacles, including unsatisfactory drug loading, disappointing active targeting, and poor stability of DDS during storage and in vivo circulation, have limited their clinical applications. Therefore, it is imperative to design tumor-specific, stimuli-responsive DDS to improve therapeutic efficiency and reduce toxicity to healthy tissues [11, 12].
Cancer is a complex disease and tumorigenesis is a multistep process involving initiation, progression, and metastasis, which are greatly affected by the tumor microenvironment (TME) [13]. The TME is the fertile soil on which tumor cells live, comprising many types of noncancerous cells (e.g., fibroblasts, endothelial cells, immune cells, and vascular cells) that are fed by the vascular system and secrete extracellular components (e.g., cytokines, growth factors, hormones and extracellular matrix (ECM)) to support the survival of the tumor cells [14]. The TME has profound effects on therapeutic outcomes and serves as a key noncancerous target of complementary cancer treatments [15]. Furthermore, the microenvironment of tumor cells is different from that of normal cells in many ways, including possessing mesh-shaped blood vessels, an acidic pH, an abnormal redox state and a specific enzyme expression profile [16-18]. These enzymes are essential biochemical components and play a critical role in cancer development due to an unbalanced distribution of their expression/activity [19]. Some unique characteristics of these enzymes, including efficient and mild catalysis, high selectivity and substrate specificity, and strong disease association, can be utilized to render nanocarriers modified with enzyme-labile linkages as ideal, intelligent DDS to provide on-demand enzyme-responsive drug release and reduce side effects. Excellent examples of such DDS are the cathepsin-based nanoplatforms [20-24].
Cathepsins are lysosomal proteases that are widely distributed in tissues. They are divided into classes depending on their active site amino acid, specifically aspartate (two proteases), cysteine (11 proteases), and serine (two proteases) [25]. As a primary member, cathepsin B is a key cysteine protease ubiquitously expressed in all mammalian lysosomes, but it is overexpressed and activated in a variety of aggressive and metastatic tumor cells and premalignant lesions to regulate cancer progression and malignancy [26]. It has been calculated that the concentration of active cathepsin B in lysosomes of tumor cells can be as high as 1 mmol/L, constituting up to 20% of the total lysosomal protein which was assumed to be 312.5 mg/mL as calculated by Henning [27], while the content of cathepsins B in normal tissues is far lower than that in lysosomes, which is even negligible [28]. It is also considered to be an important clinical prognostic biomarker to predict therapeutic efficacy against many cancers [29]. In this regard, a series of cathepsin B-responsive nanosized drug delivery systems (nanoDDS), concurrently possessing nanoscale-mediated enhanced permeability and retention (EPR) effect and specific enzyme-induced TME recognition, have been constructed to promote tumor-specific drug release from nanocarriers for targeted monitoring and lesion ablation [30].
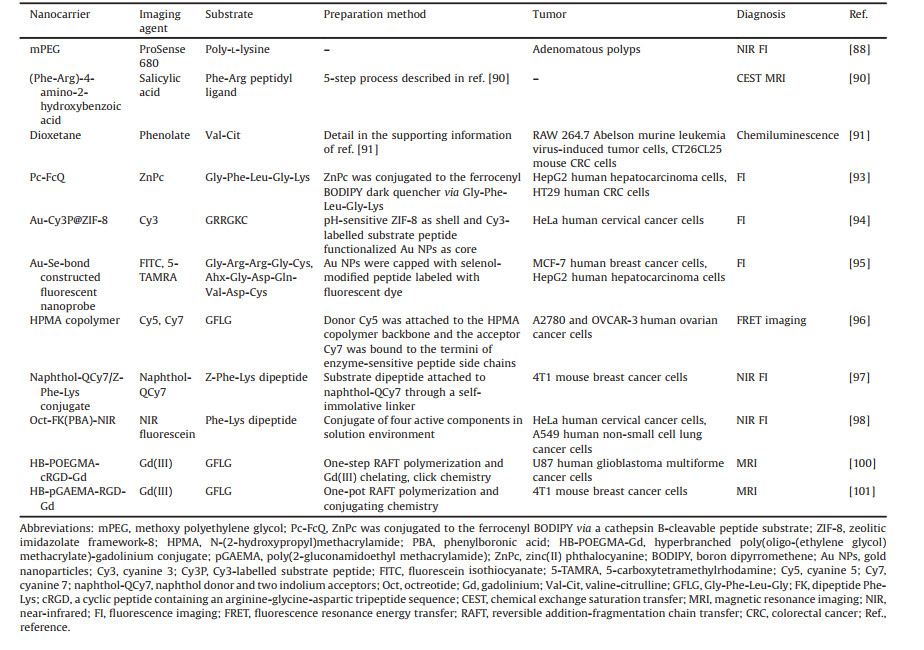
In this current review, recent advances in the design of cathepsin B-responsive DDS and their applications in controlled drug delivery are systematically summarized and analyzed. First, the structure and physiological/pathological functions of cathepsin B are described to highlight its potential as a delivery trigger. Second, multifarious cathepsin B-driven nanoDDS are investigated in categories and their progress in cancer diagnosis, treatment, and synergistic theranostics is explored. Finally, challenges and obstacles are discussed to lay the foundation to achieve the clinical application of cathepsin B-responsive DDS in the near future. The structure, function, and application framework of cathepsin B-responsive nanoDDS are outlined in Fig. 1.

|
Download:
|
| Fig. 1. The framework for the applications of cathepsin B-responsive nanoDDS in cancer diagnosis and treatment. | |
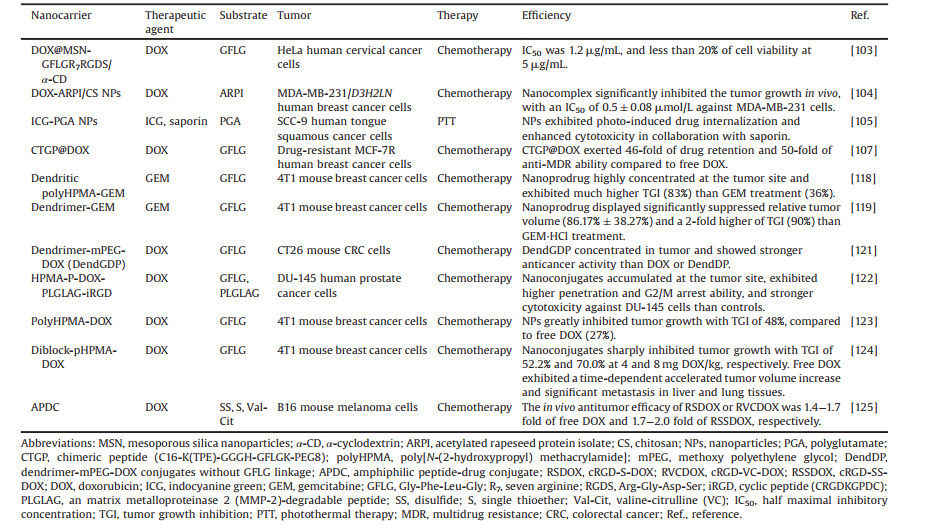
Cathepsin B (EC 3.4.22.1) is a lysosomal cysteine protease of the papain family [31]. Its main function is to degrade proteins that enter lysosomes from outside the cell or from other intracellular organelles. Cathepsin B plays a vital physiological function in maintaining cellular homeostasis by regulating many processes, including digestion, adipogenesis, hormone liberation, peptide synthesis, and immune responses. However, its altered expression and distribution can, in turn, contribute to its pathological function in mediating multiple diseases, including inflammation, infection, cardiovascular disorders, autoimmune diseases, neural degeneration, and cancer. In this section, the structure and function of cathepsin B are summarized in Fig. 2 and thoroughly dissected in order to provide a basis for its role as both responder and target in disease diagnosis and treatment, focusing on cancer.

|
Download:
|
| Fig. 2. Different functions resulting from structurally related activity of cathepsin B and their regulatory strategies. | |
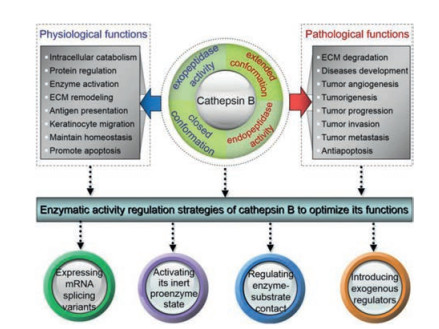
Cathepsin B (IUBMB classification: EC 3.4.22.1) is encoded by a 25.7 kb gene, located on the short arm of chromosome 8 (8p22-p23.1) and containing 12 exons [32]. After removing the internal dipeptide from single-chain cathepsin B (31 kDa), mature cathepsin B is formed, consisting of a light chain (5 kDa) and heavy chain (26 kDa) that are connected by disulfide bonds [33]. This structure consists of two separate domains that interact through an extended and soluble v-shaped polar interface. There are six disulfide bridges and two unpaired cysteine residues, Cys29 and Cys240. The active site Cys29from the left domain and a histidine residue (His199) from the right domain interact to form an ion pair due to their thiol and imidazole side-chains, which catalyze peptide-bond cleavage over the pH range of 4.0-8.5 [34]. Peptide bond cleavage is catalyzed by the nucleophilic attack of S- of Cys29 on the substrate carbonyl carbon atom, followed by proton donation from His199. The specific pockets S3, S2, S1, S1', and S2' are formed by loops in the left (S3, S1, S2') and right (S2, S1') lobe [33].
Cathepsin B possesses a unique structural element, the occluding loop containing 20 amino acids (Ile105-Thr125), which endows it with both endopeptidase and exopeptidase activity and distinguishes it from other cysteine cathepsins. In the open conformation caused by pH change or inhibitor binding, two salt bridges between the loop and enzyme (His110-Asp22 and Arg116- Asp224) are disrupted and large substrates can be bound and cleaved internally, imparting cathepsin B with endopeptidase activity [35]. However, in the closed conformation, the occluding loop binds to the enzyme via the two salt bridges and blocks the primed binding region, causing closure of the active site cleft and preventing large substrates from binding to the S2' and S3' subsites. Simultaneously, the occluding loop endows cathepsin B with dipeptidyl carboxypeptidase activity. The polar interaction between two protonated histidine residues (His110 and His111), which are located at the top of the occluding loop, and the Cterminal carboxyl group of the peptide substrate, contributes to the exopeptidase (peptidyl dipeptidase) activity of cathepsin B [36]. When twelve residues in the occluding loop are deleted, a mature cathepsin B mutant is formed, which has been shown to possess endopeptidase but not exopeptidase activity [35].
Mechanistically, cathepsin B hydrolyzes peptide bonds following anacyl-transfer reaction, which is initiated by the nucleophilic attack of the deprotonated active site cysteine residue, forming an acylenzyme intermediate that is subsequently hydrolyzed. This mechanism provides a reference for the molecular design of specific cathepsin B inhibitors. Most of these inhibitors contain electrophilic modules that bind them covalently to the sulfur of the active site cysteine, or they contain peptide fragments that noncovalently interact with the binding pockets of cathepsin B [37].
To summarize, cathepsin B has a unique molecular structure that differentiates it from other enzymes, and it shows different proteolytic activities in different folding states. These characteristics allow it to perform the corresponding biological functions in different microenvironments.
2.2. Cathepsin B functionThe multiple enzyme activities of cathepsin B determine its differential biological functions. The exopeptidase activity of cathepsin B influenced by its closed conformation is dominant in the acidic lysosomal cavity and this physiological function of the enzyme is especially important in intracellular catabolism. The endopeptidase activity of cathepsin B associated with its extended conformation prefers the neutral extracellular environment and governs pathological processes such as degradation of the ECM. Both activities have been shown to participate in tumor progression, while the endopeptidase activity plays a major role in tumor invasion and metastasis [38-40].
As cathepsin B is mainly distributed in lysosomes, its proteinregulatory role within this organelle has been the overwhelming focus of previous reports [41]. Clearly, cathepsin B plays a key role in a variety of specific physiological events, for example enzyme activation, ECM remodeling, central nervous system (CNS) homeostasis, antigen presentation, and keratinocyte migration [42-45]. On the contrary, fluctuations in the level, distribution, and activity of cathepsin B also contribute to the development of various diseases including Alzheimer's disease, virus infections, pancreatitis, rheumatoid arthritis, and especially cancer [46-50].
The activity of cathepsin B greatly affects its function; thus, it is necessary to optimize its in vivo behaviors by regulating its enzymatic activity. There are several regulatory options worth considering, which include expressing mRNA splicing variants that encode truncated cathepsinB to regulateits cellular localization and alter biofunctionality [51], activating the inert proenzyme state of cathepsin B [52], regulating enzyme-substrate contact most likely by changing its endosomal/lysosomal distribution, adjusting its cellular localization, optimizing the balance between cathepsin B and its endogenous inhibitor [53], and introducing exogenous molecules that can regulate cathepsin B expression [37].
According to the above, cathepsin B possesses molecular structure-dependent physiological/pathological functions, which can be improved by a series of activity regulation programs. Furthermore, the interaction between cathepsin B and disease development/progression, especially with cancer, and the multiple roles of cathepsin B in disease management need to be clarified.
2.3. Cathepsin B and cancerCancer is a complex disease that consists of a network of interconnected signaling pathways and complicated microenvironments [54]. The TME is a hotbed for supporting tumor growth, proliferation, invasion, and metastasis, and for encouraging the immune escape of tumor cells. Cathepsin B is a key lysosomal acidic protease that is highly activated in a variety of cancerous and preneoplastic cells due to gene amplification, enhanced transcriptional efficiency, and the presence of more stable and easily translated mRNA variants [55]. Cathepsin B plays an indispensable role in degradation of the ECM and basal lamina, highlighting its importance in tumorigenesis, progression, and metastasis. In clinical practice, cathepsin B is also considered to be a prognostic biomarker for tumor progression and therapeutic outcomes because of its predictive capacity for tumor size, tumor grade, growth factor levels, and lymph node diffusion [56].
Cathepsin B is also important in tumor angiogenesis, the formation of new blood vessels to promote the growth and metastasis of primary tumors. The process of tumor angiogenesis includes ECM degradation and the subsequent proliferation and migration of endothelial cells. Cathepsin B promotes ECM degradation and angiogenesis either by direct enzymatic activity or by activating the more powerful degradative enzymes, matrix metalloproteinases (MMPs) [57]. With degradation of the ECM, some growth factors that are bound to it, such as vascular endothelial growth factor (VEGF), are released to further promote tumor angiogenesis [58].
In addition, cathepsin B is closely associated with cell apoptosis, which is shown to maintain homeostasis and clear damaged or senescent cells. Normal apoptosis can inhibit tumor progression, but defects in the apoptosis pathway significantly improve the survival and growth of tumor cells. Cathepsin B plays a doubleedged role in the regulation of apoptosis. It has an antiapoptotic effect by inactivating Bak, activating Mcl-1, and activating caspase inhibitors. Conversely, it can activate Bid, which induces the release of cytochrome c from the mitochondria into the cytoplasm, activating the caspase family to promote apoptosis [59].
Briefly, the relationship between cathepsin B and cancer has been described and the conclusionis that cathepsinB plays a crucial rolein maintaining homeostasis, mediating cell invasion and metastasis, promoting angiogenesis, and regulating ECM remodeling. On one hand, it can be countered as a potential therapeutic target by designing appropriate inhibitors. On the otherhand, it canbe used as an activator for microenvironment-recognizable cancer management by identifying specific enzyme substrates and then linking them to bioactive molecules. In view of this, the latest progress in the constructionof cathepsin B-responsive DDS and theirapplications in the precise diagnosis, targeted therapy, and collaborative theranostics of malignant tumors is an important academic resource and will be detailed in the following sections.
3. Cathepsin B-responsive nanoDDS for precise diagnosis of malignant tumorsHighly sensitive and precise diagnosis is an important prerequisite for targeted cancer therapy. Effective imaging methods play a key role in viewing tumors and tracking their development. Commonly used cancer detection methods in the clinic include ultrasonography, general X-ray examination, computed tomography (CT), magnetic resonance imaging (MRI), and fluorescence imaging (FI). These diagnostic methods have advantages such as universality, low costs, negligible radiation damage, high sensitivity, and multi-dimensional imaging. However, traditional diagnostic methods based on molecular contrast agents suffer from molecular leakage and tissue toxicity in vivo, resulting in blurred imaging results. To overcome these limitations, a variety of nanotechnology-based tumor-specific, enzyme-responsive DDS have been identified and provide great opportunities for the early detection of cancer [60-62].
3.1. Cathepsin B-mediated cancer diagnostic procedure and representative preparation strategies for nanocarriersCathepsin B-mediated cancer diagnosis is primarily accomplished by the following steps: 1) designing a novel and highsensitivity imaging contrast agent, 2) attaching a cathepsin Bcleavable peptide sequence, 3) injecting the nanosized imaging molecules into the circulatory system through an optimal administration route, 4) cathepsin B that is highly expressed in the tumor site cleaves the substrate peptides according to the designed system, and 5) the active contrast agent is released to achieve high signal-to-noise ratio imaging. As a result, the tumor is visualized and the tumor/normal tissue boundary is clearly delineated, allowing the clinician to completely remove the tumor.
Accurate quantification, distribution, and bioactivity of enzymes are essential to intelligently guide imaging agents to track tumor progression and therapeutic efficiency. In recent years, a number of studies have clarified the localization, content and biopotency of cathepsin B by virtue of multiple imaging modalities, based on the success of various forms of multifunctional nanocarriers [63], such as liposomes [64], polymeric nanoparticles (NPs) [65], micelles [66], dendrimers [67], inorganic NPs [68, 69], quantum dots [70], nanocantilever [71]. Representative progress was summarized in Table 1.
|
|
Table 1 The latest progress of cathepsin B-responsive nanoDDS and their applications in the precise diagnosis of malignant tumors. |
The size, shape, surface characteristics, and targeting ligands of the drug-loading NPs play key roles in their biodistribution and efficacy. Observably, the well-designed NPs (10-100 nm) can be localized into the tumor sites and blocked by the normal vasculature (requires sizes of 1-2 nm) [72]. The morphology of NPs has important effects on their internalization mechanism and in vivo circulation. Previous report has revealed that non-spherical nanostructures exhibit completely different internalization modes and speeds with respect to their different initial contact angles with target cells [73]. Surface properties of nanosystems, especially surface charges, are crucial in determining their internal behaviors. NPs with slightly positive or negative charges have less interaction with each other, whereas strongly charged particles are readily cleared by macrophages in the reticuloendothelial system. Negatively charged NPs are more likely to be rejected due to the electronegativity of blood vessels and cell surfaces. Therefore, surface charge regulation is an effective strategy to enrich nanoDDS in tumor focus [74]. These physicochemical properties of nanoDDS greatly affect their biological functions and can be controlled by screening reasonable preparation techniques.
The commonly used nanoscale construction strategies are divided into three pathways, namely physical, chemical, and biological approaches. The conventional physical methods are gasphase deposition [75] and electron beam lithography [76]. Chemical methods are the most widely adopted strategies for preparing stable and uniform cargo-loading nanosystems, mainly include coprecipitation [77], redox process [78], thermal decomposition [79], hydrothermal [80] and solvothermal [81], microemulsion [82], sol-gel [83], and so on. The biological approach is a simple and ecofriendly method for the preparation of highly stable and controlled nanodrugs assisted by biomass [84]. All of which give nanoDDS the desired performance and functionality for biomedical applications. In addition to the classical preparation methods, several new high-throughput techniques have also been developed for the mass preparation of uniform and controllable nanoagents, typically listed below.
Glangchai et al. fabricated cathepsin B-responsive, biodegradable NPs with precisely controlled size (< 50 nm), morphology, and composition, using a step and flash imprint lithography (S-FIL) technique. The NPs were mass-produced on silicon wafers and were easily harvested in aqueous media. Fluorescently labeled antibodies or proteins were used as model drugs encapsulated in enzymatically degradable vectors containing acrylated peptides and acrylated polyethylene glycol macromers during the nanoprinting and etching process. The entrapped contents were accurately and controllably released in response to specific physiological or pathophysiological conditions, but still possessed nondestructive bioactivity. This high-throughput nanofabrication technique provides a potential strategy for the mass preparation of homogeneous and controllable nanomaterials for the cathepsin Btriggered, targeted delivery of active pharmaceuticals or imaging agents [85]. Bellat et al. constructed two new types of nanotransformers (NTFs), NTF1 and NTF2. After being activated by cathepsin B in lysosomes, these NTFs underwent different biomechanical remodeling to produce degradation and aggregation effects, respectively. These properties correspondingly promoted or delayed payload release into the tumor cells to exhibit tumorcell killing that was comparable or weaker, respectively, than that of the free drug. These novel NTFs with enzyme-inducible molecular structure transformation and payload release behavior were shown to be ideal nanocarriers for active drugs and contrast agents [86].
In a word, a variety of carriers can be selected to construct multifunctional nanoplatforms for cancer diagnosis and treatment by adopting optimized nanostrategies. Noteworthily, the biocompatibility of carrier materials and their compatibility with bioactive components, the efficiency and economy of the preparation process, the effect of physicochemical properties of nanocomplexes on their clinical efficacy, the circulation of nanoDDS in vivo, and so on, are important factors to determine the theranostic activity of nanoagents and need to be strictly considered in system design and preparation.
3.2. Assays for the activity and content of cathepsin BThe content and activity of enzymes are crucial to their biological functions. Therefore, real-time quantitative measurement of protease activity in living cells is an important prerequisite to explore the physiological/pathological functions of proteases and to use them as indicators of disease progression and therapeutic efficacy. Common methods for detecting the level and activity of cathepsin B include molecular biology approaches, FI, MRI, radiotracer techniques, and other highly sensitive detection methods such as chemiluminescence [87]. In a previous study, inducible near infrared (NIR) fluorescent probes were designed and utilized to simultaneously image independent biological activities associated with tumor progression in real time. This study revealed a dynamic relationship between cathepsin B activity and tumor-related inflammatory processes. It also highlighted the importance of live imaging of cathepsin B activity in illuminating polyp growth/regression dynamics and predicting tumor development and therapeutic response [88]. Analogously, an NIR-labeled activity-based probe (GB123) with an acyloxymethylketone warhead was synthesized and used to identify and localize activated cathepsin B in cerulein-induced pancreatitis. With reflectance imaging and two-photon confocal microscopy of intact organs, significantly elevated expression of activated cathepsin B was found in the inflamed mouse pancreas and in pancreatic fluid from patients with chronic pancreatitis, which could be reversed by cathepsin inhibition. This study revealed the causal role of active cathepsin B in pancreatitis and pain, and the importance of activity-based probes in indicating disease progression and therapeutic reaction [89]. Hingorani et al. designed a single diamagnetic agent with enzyme-responsive (dipeptidyl ligand) and enzyme-unresponsive (salicylic acid moiety) chemical exchange saturation transfer (CEST) signals. Concentration-independent measurements of cathepsin B activity were performed by comparing the ratio of these signals. High sensitivity, low toxicity, and molecular architecture flexibility facilitated the clinical translation of this series of extended imaging agents and this detection technique for quantifying enzyme activity [90].
In order to further improve the sensitivity of protease activity detection, Roth-Konforti et al. pioneered the use of chemiluminescence to quantify endogenously expressed cathepsin B activity under physiological conditions. The first cathepsin B chemiluminescence probe, with a triggering group consisting of a cathepsin B-cleavable peptide (valine-citrulline, Val-Cit), a self-immolative linker, and a variable motif, was designed to evaluate enzyme activity. After cleavage of the R1-Val-Cit moiety, the aniline linker underwent spontaneous 1, 6-elimination to form a phenolate, which was chemiexcited to emit green fluorescence efficiently in the physical environment. Using this probe and technique, cathepsin B was successfully imaged and quantified in leukemia and colorectal cancer (CRC). This concept could be extended to construct a variety of analogous probes and be used for the broader detection of tumor-related protease biomarkers [91]. Furthermore, Ryan et al. linked a bright-green chemiluminescent scaffold to the substrate dipeptide of cathepsin B (Val-Cit) and quantified enzyme activity via the abovementioned chemiluminescence mechanism. Through performance-driven molecular modification and structural optimization, several derivatives were synthesized and compared with cathepsin B imaging. It was found that the chemiluminescence intensity increased 18 times after connecting an acrylate ester to the chemiluminescent core. When the dipeptide recognition motif was added to a trivalent tumor targeting peptide sequence (CGKRK), the resulting molecule had better solubility and cellular absorption, resulting in a further 4.5- fold increase in chemiluminescent efficiency, far greater than other competitor candidates [92].
Equally important as or more important than accurate quantification of enzyme activity, cathepsin B has also been increasingly used as a responder to guide imaging agents in visualizing tumor development and therapeutic responses. In a variety of diagnostic models, highly sensitive FI- and clinically popular MRI-related cathepsin B-responsive nanoscale imaging systems are discussed emphatically in this section.
3.3. Cathepsin B-responsive nanoDDS for fluorescence imaging-based cancer diagnosisThe high sensitivity and flexible molecular maneuverability of FI make it an advantageous choice for this purpose. A smart cathepsin B-responsive fluorescent probe, Pc-FcQ, was developed using ferrocenyl boron dipyrromethene (BODIPY) as the dark quencher and zinc(II) phthalocyanine (ZnPc) as the fluorescent unit, which was conjugated to the quencher via a cathepsin Bcleavable peptide substrate (Gly-Phe-Leu-Gly-Lys). In the original molecular state, the fluorescence of Pc-FcQ was quenched through energy transfer to the BODIPY and subsequently the photoinduced electron transfer from the nearby ferrocenyl unit. After being recognized by cathepsin B, the peptide bonds of the substrate were cleaved, followed by the separation of ZnPc and ferrocenyl BODIPY. Subsequently, the fluorescence emission of Pc-FcQ was restored and singlet oxygen generated in HepG2 cells and tumor-bearing nude mice, with fluorescence intensity gradually increasing at the tumor site over 10 h. The results showed that Pc-FcQ was a cathepsin B-responsive fluorescent probe for tumor imaging [93].
In another report, an elaborate core-shell NP@metal-organic framework (MOF) nanoprobe, Au-Cy3P@ZIF-8, with a pHresponsive zeolitic imidazolate framework-8 (ZIF-8) as the shell and a cyanine 3 (Cy3)-labeled substrate peptide (Cy3-GRRGKC) functionalized gold NP (Au-Cy3P) as the core, was fabricated to sense an acidic TME and cathepsin B, respectively. After internalization into tumor cells, the ZIF-8 shell decomposed in response to the acidic environment, exposing the substrate peptide, which was recognized by cathepsin B to cleave the Cy3-labeled moiety on the AuNP surface and illuminate cells. This dual-responsive FI strategy could specifically highlight lysosomal cathepsin B (pH 4.5-6.0) to image precisely active enzymes in cells for disease surveillance [94].
In a more recent study, an Au-Se-bonded nanoprobe, in which Au NPs were functionalized with two different selenol-modified peptide chains labeled with two types of fluorescent dyes, which specifically targeted upstream cathepsin B and downstream caspase-3 proteins, respectively, was constructed to monitor intracellular apoptotic signal transduction in real time. Upon stimulation of the apoptotic pathway in living cells, the sequential fluorescent signals indicated the successive activation of cathepsin B and caspase-3. This fluorescent nanoprobe, having good stability and high fluorescence efficiency, demonstrated potential to monitor the interactions of signaling molecules in vivo for the pathological analysis of cancer [95].
Because of the high penetration of organic tissues by NIR photons and low tissue autofluorescence at NIR wavelengths, various NIR fluorescent probe-based enzyme-responsive DDS have been designed and used for targeted diagnosis of tumors. Zhang et al. designed an NIR fluorescence resonance energy transfer (FRET) system, in which the donor Cy5 was attached to the main chain N-(2-hydroxypropyl)methacrylamide (HPMA) copolymer backbone and the acceptor Cy7 was conjugated at the end of an enzyme-sensitive oligopeptide (Gly-Phe-Leu-Gly, GFLG) side chain with optimal relative positioning (< 10 nm). When the substrate peptide was specifically recognized by cathepsin B in tumor cells, the bond between the peptide and Cy7 was cleaved and FRET was eliminated, resulting in the effective release and high concentration of payloads inside tumors to achieve tumor diagnosis and analyze the in vivo behavior of imaging agents [96].
After molecular modification, two novel cathepsin B-inducible NIR fluorescent probes with different activation mechanisms (i.e., internal charge transfer (ICT) and FRET, respectively) were synthesized and evaluated for FI potential. It was found that the "OFF state" fluorescence intensity of the ICT probe was significantly lower than that of the FRET probe, symbolizing a higher signal-tonoise ratio, sensitivity, and contrast capability of the former to monitor tumor progression [97].
In a recent study, a sophisticated triple-targeting NIR fluorogenic probe, Oct-FK(PBA)-NIR consisting of octreotide (Oct) as a synthetic ligand of somatostatin receptors (SSTRs) for the first target, a H2O2-activatable phenylboronic acid (PBA) for the second target, a dipeptide substrate of cathepsin B (Phe-Lys (FK)) for the third target, and an NIR fluorophore, was synthesized and used for multi-stimulated, selective NIR imaging of cancer cells without visualizing normal cells [98].
Given the excellent luminescence sensitivity and stability of fluorescent materials, a variety of fluorescent cores such as organic small molecules, macromolecules, inorganic or endogenous fluorophores, have been integrated into a wide range of multifunctional nanocomposites for tumor-specific lighting (Fig. 3) [99]. However, the biocompatibility, fluorescence efficiency and lifetime of novel luminescent materials are the prerequisites that must be considered for cancer diagnosis.

|
Download:
|
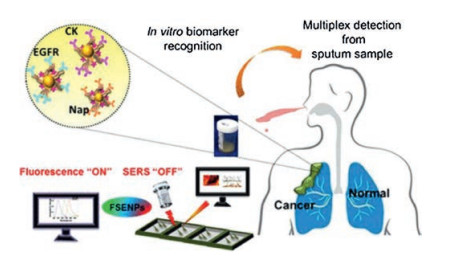
| Fig. 3. Enzyme-responsive switchable fluorescence-SERS diagnostic nanocomplexes were used in the multimode detection of lung cancer biomarkers. SERS, surface enhanced Raman scattering; EGFR, epidermal growth factor receptor; CK, cytokeratin-19; Nap, napsin-A; FSENPs, fluorescence-SERS encoded nanoparticle probes. Reproduced with permission [99]. Copyright 2018, American Chemical Society. | |
MRI is a noninvasive technique widely used in the clinic for tumor diagnosis and treatment efficacy monitoring because of its high spatial resolution, quantitative 3D imaging, and pathological assessment competence. In recent years, the construction and application of enzyme-induced nanoMRI systems has attracted extensive research interest. Accordingly, a well-defined and sizeadjustable hyperbranched polymer-gadolinium (Gd) conjugate, HBPOEGMA-cRGD-Gd, was prepared via one-step polymerization, Gd(III) chelating and click chemistry. Upon exposure to cathepsin B, the high molecular weight (MW) (180 and 210 kDa) and nanoscale-sized (38 and 42 nm) complex conjugates were degraded into low MW (25 and 30 kDa) and smaller-sized (4.8 and 5.2 nm) fragments. The MRI effect of this nanocomplex was three times more powerful than that of clinical diethylenediami-nepentaacetic acid (DTPA)-Gd, and there were almost no side effects on U87 cells and tumor models, demonstrating its great potency as a new generation of MRI contrast agent for enzymeresponsive cancer diagnosis [100].
Furthermore, two TME-responsive branched Gd-based glycopolymer conjugates were synthesized by one-pot polymerization and conjugating chemistry. A branched polymer connected to the GFLG oligopeptide was conjugated with 1, 4, 7, 10-tetraazacyclodo-decane-1, 4, 7, 10-tetraacetic acid (DOTA)-Gd to generate a cathepsin B-sensitive MRI contrast agent with a MW of 124 kDa. Its targeted homolog with a MW of 136 kDa was prepared by introducing the tumor-targeting cRGDyK cyclic peptide to the branched molecule. Both agents had tunable molecular and nanoscale structures and superior MRI performance, with an aqueous phase relaxation efficiency of 12.3 and 13.2 L mmol-1 s-1, respectively, four times higher than that of DTPA-Gd (2.9 L mmol-1 s-1). The significantly enhanced MRI signal intensities and negligible in vitro and in vivo toxicities identified these branched macromolecules as intelligently responsive tumor-targeting imaging agents [101].
To summarize, nanocarrier-based multifunctional complexes loaded with novel imaging agents and cathepsin B-cleavable substrates have received significant attention as TME-responsive contrast agents for highly sensitive cancer diagnosis. Despite great progress in the preparation and applications of these imaging systems, there remain controversies regarding the molecular design, system composition, and material safety of these novel multifunctional contrast agents.
4. Cathepsin B-responsive nanoDDS for targeted therapy of malignant tumorsCathepsin B-related cancer treatment mainly utilizes three schemes: directly impede cathepsin B activity using endogenous inhibitors, synthetic inhibitors, or antisense cDNA plasmid; constructing nanocomposites containing unmodified drugs and enzyme-responsive substrate sequences; and synthesizing and nanosizing the prodrugs of the drug-vehicle-enzyme-responsive sequence complex. Among them, the strategy of inhibiting cathepsin B activity is beyond the scope of this paper. The other two nanotherapeutic-based drug delivery strategies will be discussed in detail below.
In recent years, research has focused on the construction of multifunctional nanocomposites and their applications in improving the therapeutic effect on tumors. Nevertheless, only a few nanoDDS have been approved for clinical use, mainly due to unwanted drug leakage during in vivo transport and poor tumor targeting [102]. Cathepsin B has been shown to be an important factor in mediating the formation, growth, invasion, and metastasis of a wide variety of tumors including melanoma, glioma, breast cancer, CRC, gastric cancer, lung cancer and ovarian cancer. Considering all of the above reasons, various cathepsin Bresponsive nanoplatforms loaded with active pharmaceutical or novel prodrugs have been widely studied for TME-targeted cancer treatment. Representative research advances are systematically summarized in Table 2 and analyzed in this section.
|
|
Table 2 Illustrative examples of cathepsin B-responsive nanoDDS and their applications in the targeted therapy of malignant tumors. |
In order to reduce the side effects of the chemotherapeutic drug doxorubicin (DOX) and improve its tumor targeting, rotaxanemodified mesoporous silica nanoparticles (MSNs) were designed to enhance its therapeutic efficiency. The classic rotaxane was anchored onto the orifices of MSNs as a gatekeeper and fabricated by using an alkoxysilane tether, α-cyclodextrin (α-CD), and multifunctional peptides, which consisted of a heptaarginine (R7) cell-penetrating peptide, a cathepsin B-cleavable peptide (GFLG), and a tumor-targeting peptide (RGDS). DOX was sealed into the nanopores of the MSNs and the drug-loaded nanoplatforms were targeted to tumor cells, efficiently permeating cell membranes and exhibiting an "off-on" drug release behavior. This resulted in an 80% release rate and promoted apoptosis of αvβ3-positive HeLa cancer cells [103].
In a collaborative medication system, dual acidic pH- and cathepsin B-responsive multidrug synergistic nanocomplexes, DOX-ARPI/CS NPs, were prepared using an ionic self-assembly technique. Acetylated rapeseed protein isolate (ARPI), a derivative of the biocompatible protein isolates of rapeseed, is rich in carboxyl (COOH) and amino (NH) groups on its molecular skeleton for increased solubility and pH-buffering capability. ARPI can be catalytically degraded by cathepsin B into peptides that are antiproliferative against cancer cells. When anionic ARPI was used as a nanocarrier and combined with the cationic polymer chitosan (CS) or cationic DOX via an ionic self-assembly process, the resulting nanocomplexes responded to intracellular acidic pH and lysosomal cathepsin B, leading to the release of anticancer peptides into the cytosol and enhanced DOX trafficking to the nucleus. Ultimately, this synergistically enhanced drug delivery and cytotoxicity [104].
Another synergistically enhanced, multi-mechanism medication system was developed by combining NIR photosensitizing indocyanine green (ICG)-loaded polyglutamate (PGA) NPs with a ribosome-inactivating protein (saporin). This nanocomplex was degraded by cathepsin B, which resulted in decreased particle size, increased intracellular ICG concentration, and time-dependent NIR luminescence. Under NIR irradiation, the nanocomposites exhibited facilitated saporin release from lysosomes and synergistically enhanced antineoplastic activity, demonstrating great potential for the enzyme-responsive treatment of superficial tumors [105].
Addressing the problem of drug resistance [106], an ingenious multifunctional nanocarrier, a chimeric peptide C16-K(TPE)- GGGH-GFLGK-PEG8 (CTGP) with cathepsin B-responsive and cell membrane-targeting dual function, was designed and selfassembled into nanomicelles for DOX loading to develop the novel nanodrug CTGP@DOX. After administration, pericellular-overexpressed cathepsin B catalyzed the cleavage of the GFLG sequence, resulting in the decomposition of CTGP@DOX and its morphological transformation from NPs to nanofibers due to hydrophilic-hydrophobic conversion and hydrogen bonding. The one-dimensional nanofiber could target cell membranes and tightly adhered to and wrapped around cells to prevent DOX efflux. This perfectly constructed nanotherapeutic showed great potential to reverse drug resistance, with 46 times the DOX retention and 50 times the anti-resistance capacity in vitro and in vivo of free DOX. This represented a novel tumor treatment mechanism characterized by the enzyme-responsive morphology transition-mediated reversal of drug resistance and improvement in therapeutic efficacy [107].
Beyond that, a variety of other nanoDDS based on physical loading technologies have also been developed to efficiently preserve drug potency (Fig. 4) [108-111], but the imprecise coordination of carrier and bioactive molecule proportions and the strict loading strategies limit their routine use. In view of this, quantitative coupling of carrier materials with active components and universal formulations are effective ideas to overcome these obstacles.

|
Download:
|
| Fig. 4. The high drug efficiency and mechanism of artemisinin-based smart nanomedicine (Tf-MSN@AB). ART, artemisinin; ROS, reactive oxygen species; GSH, glutathione; Tf, transferrin; MSN, mesoporous silica nanoparticle. Reproduced with permission [110]. Copyright 2019, American Chemical Society. | |
Prodrugs, known as chemically coupled bioinert compounds that are metabolized in vivo and reproduce the parent bioactive components [112]. They have overwhelming advantages such as enhanced water solubility and membrane permeability, exact component ratio, improved cell uptake, decreased adverse effects, and flexibility in modification of targeted or intelligent response modules such as cathepsin B [113-115]. Prodrugs can effectively overcome the low solubility, poor absorption, undesirable half-life, and multidrug resistance of free drugs, as well as the defects of nanoDDS based on non-covalent physical encapsulation, such as poor stability, undesired cargo leakage during circulation, and obvious batch-to-batch differences in drug loading and release kinetics, especially for nanosystems composed of components with different physicochemical properties [116, 117].
Inspired by the successful development of various intelligent nanoDDS, conjugating therapeutic drugs with stimuli-responsive molecular switches and formulating them to nanoscale levels has attracted extensive research interest in spatiotemporal-controlled cargo delivery for the targeted therapy of tumors. Cathepsin B, as an important responder overexpressed in a variety of tumor cells, has been introduced into multifarious enzyme-sensitive nanoDDS by conjugating active pharmaceuticals to the carriers via a zymolyte linkage. In addition, an appropriate addition of targeting ligand may promote specific enrichment of drug in tumors. When encountered cathepsin B, the substrates in the nanocomposites are specifically identified and cleaved to release the active drugs to effectively treat cancer with decreased side effects, especially for poorly soluble and permeable but highly potent therapeutic agents.
In a representative study, an enzyme-inductive dendritic polyHPMA-GEM (gemcitabine) prodrug was synthesized in a one-pot reaction under mild conditions and nano-formulated as stable DDS with an average size of 46 nm. Upon exposure to cathepsin B, more than 95% of the GEM was released within 3 h of incubation and showed a significantly enhanced in vivo efficiency compared to that of free GEM, with tumor inhibition rates (TGIs) of 83% and 36% respectively. This increased efficacy was caused by enhanced cellular uptake, effective tumor site accumulation, and reinforced antiangiogenesis-mediated apoptosis [118]. Another similar cathepsin B-susceptible polyethylene glycol (PEG)ylated lysine peptide dendrimer-GEM conjugate was developed via an efficient click reaction. The GFLG peptide was cleaved by cathepsin B, leading to 80% GEM release after 24 h and superior in vivo antitumor efficacy in a 4T1 murine breast cancer model, with a TGI (~90%) that was twice that of free GEM [119].
For the past few years, as an effective cytotoxic anticancer drug, DOX-based enzyme-responsive conjugate precursors have also been extensively studied. In a previous report, cathepsin B-cleavable DOX prodrugs were discussed in terms of their design strategies, coupling process, antitumor efficacy, and molecular mechanism [120]. Under the guidance of this theory, GFLG-linked dendrimer-methoxy PEG (mPEG)-DOX conjugates (DendGDP) were synthesized and self-assembled into spherical NPs (< 200 nm). The nanoconjugates showed sensitive cathepsin Bresponsive drug release, tumor site drug enrichment, and significant tumor inhibition of CT26 tumor xenografts [121]. Further, a well-defined cathepsin B and MMP-2 dual-responsive HPMA-DOX-tumor penetrating peptide (CRGDKGPDC, iRGD) conjugate was developed, which was highly enriched in and permeable to monolayers and multilayered DU-145 prostate cancer cell spheroids, resulting in crisper cell cycle G2/M arrest and stronger antitumor activity [122]. A versatile cathepsin B and pH dual-susceptive branched copolymer polyHPMA-DOX conjugate (165 kDa) was prepared by a one-pot reaction, drug conjugated, and self-assembled into NPs (102 nm) with a negatively charged surface. Upon exposure to the acidic pH and high cathepsin B-expressing environment of the tumor site, the nanoconjugates exhibited enhanced breast tumor-targeted aggregation and enhanced therapeutic effect without obvious toxicity to normal tissues [123]. In another innovative design system, a diblock backbone-degradable HPMA copolymer-DOX conjugate with a cathepsin B-responsive GFLG sequence and pH-sensitive hydrazone bond was prepared via polymerization and conjugation. After administration, the acidic pH and cathepsin B overexpression in tumor cells promoted the degradation of the high-MW nanoconjugates (94 kDa) to low-MW nanoconjugates (45 kDa) and the accumulation of DOX at the tumor site, resulting in obviously superior pharmacokinetic properties, biosecurity, and antitumor activity against 4T1 xenografts compared to that of the free drug [124].
To elucidate the relationship between molecular structure and biological activity, especially the possible synergistic effect between the ligand peptides and stimuli-responsive linkages in amphiphilic peptide-drug conjugates (APDCs), three types of DOXcontaining APDCs were synthesized and their in vivo and in vitro biological functions were analyzed in parallel. It was demonstrated that a noncleavable single thioether (S) or cathepsin B cleavable valine-citrulline dipeptide (VC)-linked APDC exhibited stronger antitumor activity than free DOX (1.4-1.7 fold) and a reductioncleavable disulfide (SS)-linked APDC (1.7-2.0 fold), and superior biocompatibility. This finding provided theoretical reference and guidance for the design of ideal nanoconjugate prodrugs for effective cancer treatment [125].
In recent years, significant efforts have been made to develop diverse peptide-drug conjugates by molecular construction strategies to improve drug efficacy and reduce drug toxicity [126]. Many investigators have concluded that cathepsin Bcleavable prodrugs are less active in vitro and more effective in vivo, demonstrating the vital function of cathepsin B. However, there are still multiple challenges and obstacles in achieving clinical translation, such as the lack of in-depth mechanistic analyses and activity evaluations, undefined subcellular distributions, and lack of understanding of the prodrug release pathways. In future work when exploring novel nanoconjugates, more effort should be placed on the in-depth performance evaluation and clinical application development of existing prodrugs. Equally important, it is urgent to design new types of integrated nanoDDS co-loaded with diagnostic and therapeutic modules to realize cooperative tumor theranostics.
5. Cathepsin B-responsive nanoDDS for synergistic theranostics of malignant tumorsMultifunctional nanoprobes loaded with imaging and therapeutic components are an important approach to achieve personalized and precise diagnosis and treatment of tumors. Combined with tumor-associated enzyme recognition, various TME-targeted nanohybrids have been developed and have shown considerable efficacy in cancer theranostics (summarized in Table 3). Corresponding to the aforementioned applications of nanoagents in cancer diagnosis, only FI- and MRI-mediated collaborative cancer diagnosis and treatment are emphasized in this section.
|
|
Table 3 Recent advances in cathepsin B-responsive nanoDDS and their applications in the synergistic theranostics of malignant tumors. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
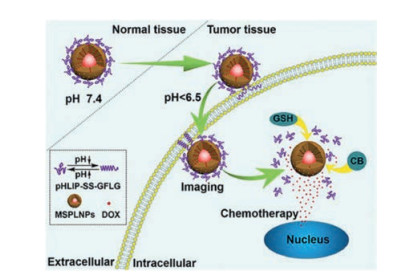
As a typical example, an enzyme-activatable, cell-penetrating peptide (CPP)-linked, quantum dots (QDs)- and DOX-loaded mesoporous silica (MS) nanocomplex was designed for nucleartargeted drug delivery. When highly expressed cathepsin B was encountered at the tumor site, the substrate peptide was cleaved, further activating oligocationic TAT (CRRRQRRKKR) residues for nucleus-directed DOX release. The enriched drug concentration at the tumor site resulted in enhanced antitumor activity in both cathepsin B-expressing sensitive and resistant cells to DOX [127]. A pH-driven and cathepsin B/glutathione (GSH) dual-responsive core-shell MS nanoprobe was fabricated to encapsulate persistent luminescence nanoparticles (PLNPs) and DOX. An acid-sensitive transmembrane helix protein, pH-low-insertion-peptide (pHLIP), was grafted onto the surface of the MS via the GFLG peptide and disulfide bonds, providing a driving force to increase the EPR effect and enzyme/GSH dual-responsiveness of the nanocomposites. Based on these characteristics, the nanoprobe exhibited increased drug uptake and tumor site aggregation, sensitive and persistent NIR imaging, enhanced cell killing, and image-mediated antitumor activity (Fig. 5) [128].

|
Download:
|
| Fig. 5. Schematic diagram of synergistic persistent luminescence imaging and tumor chemotherapy with cathepsin B/GSH dual-responsive nanoprobes, pHLIPSS-GFLG-MSPLNPs@DOX. Reproduced with permission [128]. Copyright 2020, American Chemical Society. | |
{kind=link}
Integrating the strengths of aggregation-induced emission (AIE) and enzyme-responsive drug delivery, Han et al. designed a novel multifunctional nano-prodrug, TPE-GEM-RGD, in which the GEM was conjugated to the molecular skeleton via reduction-responsive disulfide bonds; tetraphenylene (TPE), possessing an AIE effect, was conjugated to the prodrug by the GFLG peptide sequence; and an RGD peptide was introduced as a targeting group. When GFLG was cleaved by cathepsin B, intracellular fluorescence increased, and high GSH concentrations led to the release of GEM to synergistically detect and inhibit pancreatic cancer progression, respectively [129]. Another system, an enzyme-sensitive albuminbased GEM DDS, HSA-GEM/IR780, was developed by conjugation of GEM to human serum albumin (HSA) via the cathepsin Bcleavable GFLG and then complexing with the NIR dye IR780. This nanoarchitecture was clustered exclusively in tumor foci and exerted long-lasting NIR FI, suppressed drug inactivation, and greatly enhanced tumor inhibition efficiency [130].
Considering primary or metastatic tumors that lack specific molecular epitopes for drug targeting, such as triple negative breast cancer (TNBC), an antibody-drug conjugate (ADC) was developed containing a single-chain antibody (scFv) against the platelet integrin GPIIb/IIIa on activated platelets in the TME, a potent chemotherapeutic microtubule inhibitor monomethyl auristatin E (MMAE), and a cathepsin B-activatable Val-Cit linker. When cyanine 7 was introduced and the conjugate was exposed to highly expressed cathepsin B in vivo, NIR FI revealed that ADC selectively localized to primary and metastatic tumors of TNBC. Moreover, it showed remarkable therapeutic activity in inhibiting the tumor growth of TNBC, CRC, fibrosarcoma, and prostate cancer, as well as preventing TNBC from metastasizing, without identifiable side effects [131].
In order to realize reactive oxygen species (ROS)-triggered, lysosomal membrane permeabilization (LMP)-mediated cell death without irradiation, a folate (FA)-functionalized nanocapsule, encapsulating artemisinin and a dual protease light-up nanoprobe, was developed to track the intracellular localization of artemisinin and its induction of apoptosis in situ. A graphene oxide (GO) nanosheet was covalently conjugated with a rhodamine B (RhB)-labeled cathepsin B substrate peptide and fluorescein (FAM)-labeled caspase-3 substrate peptide to act as the fluorescent probe (GO-RhB-FAM), in order to track FR-mediated drug endocytosis, entry into lysosomes, diffusion to the cytoplasm, and induction of apoptosis by imaging of live, tumor-bearing mice. Therefore, this multifunctional nanocapsule could exclusively image tumors, monitor lysosomal enzyme activities in real time, and promote therapeutic efficiency [132].
As a mainstream treatment model for many types of cancer, a variety of chemotherapeutics have been loaded into highly sensitive fluorescent nanohybrids modified with multiple TMEresponsive modules for luminescence-guided and -monitored cancer treatment [133, 134]. In order to further develop the biomedical applications of these constructs, material compatibility and functional synergy between different components need to be focused in order to mediate efficiency maximization.
5.2. Cathepsin B-responsive nanoDDS for fluorescence imagingsynergized cancer phototherapyAn intelligent nanoprobe based on folate receptor (FR)-targeting and cathepsin B-responsiveness was constructed via noncovalent assembly of two functional units onto the surface of GO, which acted as a delivery carrier and fluorescence quencher. Considering the functional units, FR-targeting and enzyme sensitivity were achieved using 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[folate(polyethylene glycol)-2000] (DSPEPEG2000-FA) and chlorin e6-labeled peptide (Ce6-GRRGKGGFFFF, Ce6-Pep), respectively. After endocytosis and encountering cathepsin B, the peptide was cleaved, resulting in Ce6 release and an 18-fold increase in fluorescence intensity under 660 nm excitation. Simultaneously, the induced cytotoxic singlet oxygen stimulated lysosomal cell death and the fluorescing Ce6 diffused into the cytoplasm to monitor therapeutic efficacy in situ, thereby demonstrating its theranostic potency (Fig. 6) [135]. Jeong et al. conjugated atezolizumab (a programmed death-ligand 1 (PD-L1) antibody) with mPEG and Ce6 via a cathepsin B-cleavable peptide linker to create immune checkpoint inhibitor (ICI) nanocomposites. Upon exposure to cathepsin B, Ce6 was released from the nanoagents and exhibited enzyme level-dependent fluorescence. In an animal model of CT26 CRC cell lung metastasis, the nanocomposites demonstrated superior TME-dependent tumor targeting, significantly enhanced fluorescence imaging, and antitumor efficacy [136]. Kim et al. conjugated FA with the photosensitizer chlorin e4 (Ce4) via a cathepsin B-cleavable diarginine (RR) chemical linker, to produce the smart, small-sized, and dual-targeted theranostic agent FA-RRK(Ce4)-OH (FRC). FRC was concentrated in tumors by dual-targeting of FR and cathepsin B, enabling NIR FI with a high signal-to-noise ratio under 640 nm irradiation and selective photodynamic therapy (PDT) of FRoverexpressing KB cells and xenograft tumors without tissue toxicity [137].

|
Download:
|
| Fig. 6. The process diagram of folate receptor-mediated nanoprobe delivery and cathepsin B-responsive photosensitive lysosomal cell death and real-time efficacy monitoring. Reproduced with permission [135]. Copyright 2015, American Chemical Society. | |
{kind=link}
In addition to co-loading components with separate diagnostic and therapeutic functions into a nanocomposite system, the design and nano-functionalization of a single, multipurpose molecule that combines both imaging and medicinal effects, is another effective method for the theranostics of tumors. In a previous study, a cathepsin B-cleavable fluorescent probe, CyA-P-CyB with simultaneous NIR FI and PDT properties, was prepared containing two cyanine moieties (CyA and CyB) linked by a cathepsin B-activated GFLG peptide based on a FRET mechanism. After being cleaved by cathepsin B, the fluorescence donor CyA and acceptor CyB were separated, resulting in strong fluorescence emission to visualize tumor progression and the treatment process. Meanwhile, the PDT reagent, Cy-S-Ph-NH2 with a maximum absorption at 800 nm, was released and exhibited strong NIR-induced cytotoxicity against tumor cells and xenografted tumors in mice [138].
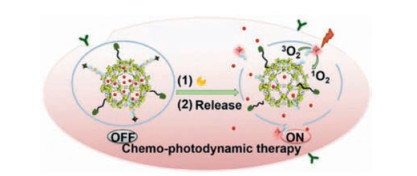
In addition, cathepsin B-responsive chemo-phototherapy has also been driven by highly sensitive FI to achieve target-specific cancer diagnosis and collaborative treatment under irradiation (Fig. 7) [139]. In the future work, nanoDDS need to be multifunctionalized under the premise of streamlining architecture but ensuring efficiency, and their internal behaviors should be clearly elucidated.

|
Download:
|
| Fig. 7. The process of enzyme-responsive cancer cell imaging-synergized chemophotodynamic therapy. Reproduced with permission [139]. Copyright 2017, American Chemical Society. | |
{kind=link}
Considering its advantages of non-invasiveness, high sensitivity, and excellent tissue penetration, MRI has played a major role in the basic research and clinical diagnosis of a variety of tumors. For instance, DOX enzyme-cleavable peptide conjugates were covalently tethered to the surface of magnetic silica nanoparticles (SMNPs) via click chemistry. The nanoconjugates were specifically degraded by highly expressed cathepsin B to present DOX selectively at the tumor location for targeted lesion suppression. Synergistically, the molecular pathway and mechanism of DOX activity were monitored visually by the combination of MRI and FI techniques [140].
Owing to the specific recognition between an enzyme and its inhibitor, selective inhibitors have been considered desirable enzyme-responsive motifs that could be introduced into nanoDDS for multipurpose tumor management. In a typical manner, a lipidated cathepsin B inhibitor NS-629 was incorporated into the envelope of a liposomal nanocarrier to form a cathepsin Bactivatable universal nanoplatform, LNC-NS-629, to carry bioactive cargoes for medical applications. When encapsulating the clinical MRI agent Magnevist (gadopentetate dimeglumine), the composite nanoprobe showed contrast enhancement at tumor sites, clear boundary outlines, and persistent signals. DOX-encapsulated LNCNS-629 was 22-fold more potent in inhibiting tumor cells than DOXnaked LNC liposomes, with respective half maximal inhibitory concentrations (IC50s) of 0.05 and 1.1 μg/mL. Furthermore, the enzyme-inhibitory activity of NS-629 provided an additional benefit for the inhibition of tumor progression and development of chemotherapy resistance [141].
A variety of other nanocomposite theranostic preparations based on optimized biocompatibility vectors, imaging agents, therapeutic agents, and intelligent response and targeting modules have also received great attention from researchers and have made encouraging progress in the coordinated diagnosis and treatment of malignant tumors [142-147]. Despite promising results, the successful preparation and theranostic application of nanocomposites still face great challenges, including complexity of the design, preparation, and functional optimization of composite systems; maintaining the performance and timeliness of active components; and the unpredictability of certain pathways of action. Hence, achieving optimal theranostic efficiency without negative consequences by introducing a minimum number of modules is the growing trend in cancer management. Future work should focus on the design, development, and nano-formulation of omnipotent molecules with multiple functions, while eliminating complex preparation techniques and extreme experimental conditions.
6. Concluding remarks and future perspectivesCathepsin B is a ubiquitously expressed human lysosomal cysteine protease and performs important physiological and pathological functions. It is necessary for cell survival, while uncontrolled regulation of its activity can also lead to many serious diseases, especially its extensive involvement in tumorigenesis, tumor progression, metastasis, angiogenesis, and apoptosis. The increasing understanding of its enzyme properties and functions makes it an important tumor diagnostic and prognostic predictor, and it has been validated as a promising therapeutic target for drug discovery. Moreover, it has been found to be a smart responder to promote the intelligent presentation and controlled release of bioactive substances.
The tremendous advantages of nanomaterials and the irreplaceable role of the TME in tumor development make enzymeresponsive, sophisticated nanoDDS a new generation of cancer theranostics. Such intelligent nanosystems offer incredible promise in overcoming the deficiencies of conventional medicines and provide great opportunities for monitoring, preventing, diagnosing, and treating malignancies, because of their sensitive TME responsiveness, spatiotemporal-controlled cargo delivery, sitespecific drug release, and enhanced accumulation in tumor lesions. To take advantage of this opportunity, various cathepsin Bresponsive multimodal nanoDDS that simultaneously incorporate imaging and therapeutic components have emerged to achieve controlled delivery of active payloads and efficient cancer theranostics [148].
Despite unprecedented and enthusiastic progress in the study of enzyme-responsive nanoDDS, meeting essential criteria for designing a clinically safe and effective nanoplatform still poses striking challenges and issues, which need to be addressed in order to realize the clinical applications of these nanoplatforms. These challenges include:
(1) Partial incompatibility between enzyme and substrate. First, in addition to the high expression in tumors, trace levels of the enzyme in blood and normal tissues induces the extratumoral cleavage of nanoDDS, resulting in reduced theranostic sensitivity and unnecessary tissue toxicity. Second, enzyme expression varies greatly with tumor location/type/progression and between patients. Third, the patterns of tumorassociated enzyme dysregulation and the spatiotemporal effect of enzyme activity have not been clearly elucidated. Beyond that, substrate-sequence overlap among closely related enzyme subtypes makes it difficult to design and screen specific zymolytes [16, 21].
(2) Inability to identify precancerous lesions and delineate tumor boundaries. Combining traditional modes with enzyme responsiveness has achieved considerable results in illuminating tumors, but sensitivity and specificity need to be improved. Hence, the ideal situation is to design highly sensitive contrast agents, especially clinical integrants with both diagnostic and therapeutic functions, and target them to the lesion by a multistimulus responsive system for high-definition imaging and synchronous treatment [149].
(3) Obstacles in the mass production of intelligent nanoDDS. Highly consistent large-scale production is an important prerequisite for the clinical applications of nanodrugs. The complexity of the preparation process needs to be optimized with emphasis on the homogeneity and stability of their biological activities and physicochemical properties, such as morphology, size, surface charge, and functional components [150].
(4) Biocompatibility and nanotoxicity. Safe and long-lasting circulation of nanomaterials in vivo is essential for their activation. The rapid development of nanotechnology has not neutralized the potential threat of biotoxicity caused by nanoscale effects. Although the mechanism of nanotoxicity has not been conclusively determined, the modification of biocompatibility and the regulation of biochemical characteristics have been shown to be desirable approaches to improve the intracorporeal efficiency of nanomaterials. Considerable progress has been made in optimizing the long-term biological impacts [151].
In conclusion, dysregulated cathepsin B expression in the TME is a critical factor in inducing tumor progression and a pivotal target for mediating tumor management by identifying specific substrates. A variety of multifunctional cathepsin B-responsive nanoDDS have been developed and have had better impacts on cancer management than traditional medicine through intelligently triggered on-demand cargo delivery, release, and activation. These sophisticated nanotheranostics, possessing applicationdriven feature addition, reasonable structure design and ingenious performance matching, have great clinical development potential. Under the premise of systematically considering carrier modification, large-scale preparation, enzyme activity, substrate specificity and degradation kinetics, they will realize clinical applications for the precise diagnosis and targeted therapy of malignant tumors.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81903662, 51903201), China Postdoctoral Science Foundation Grant (Nos. 2019M661057, 2019M653660), the Applied Basic Research Programs of Shanxi Province (No. 201901D211347), the Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (No. 2019L0428), the Startup Foundation for Doctors of Shanxi Province (No. SD1827), Natural Science Foundation of Shaanxi Province (No. 2020JQ-086), College Students' Innovation and Entrepreneurship Training Program (No. SJ201910698121), and the Startup Foundation for Doctors of Shanxi Medical University (No. XD1824).
| [1] |
R. Siegel, J. Ma, Z. Zou, A. Jemal, CA Cancer J. Clin. 64 (2014) 9-29. DOI:10.3322/caac.21208 |
| [2] |
K. Cho, X. Wang, S. Nie, et al., Clin. Cancer Res. 14 (2008) 1310-1316. DOI:10.1158/1078-0432.CCR-07-1441 |
| [3] |
Y. Xiao, F. An, J. Chen, et al., Small 15 (2019) 1903121. DOI:10.1002/smll.201903121 |
| [4] |
X. Gong, J. Zhang, S. Jiang, Chem. Commun. 56 (2020) 3054-3057. DOI:10.1039/C9CC08768K |
| [5] |
L. Zhong, X. Gong, Soft Matter 15 (2019) 9500-9506. DOI:10.1039/C9SM01624D |
| [6] |
Y. Li, Y. Yang, F. An, et al., Nanotechnology 24 (2013) 015103. DOI:10.1088/0957-4484/24/1/015103 |
| [7] |
Y. Li, Y. Chang, X. Lian, et al., J. Biomed. Nanotechnol. 14 (2018) 1515-1542. DOI:10.1166/jbn.2018.2614 |
| [8] |
Y. Li, H. Zhang, J. Biomed. Nanotechnol. 15 (2019) 1-27. DOI:10.1166/jbn.2019.2670 |
| [9] |
Y. Li, H. Zhang, Nanomedicine 14 (2019) 1493-1512. DOI:10.2217/nnm-2018-0346 |
| [10] |
Y. Li, Q. Dong, T. Mei, et al., Curr. Drug Targets 21 (2020) 228-251. DOI:10.2174/1389450120666190807143245 |
| [11] |
M. Gu, X. Wang, T.B. Toh, E.K. Chow, Drug Discov. Today 23 (2018) 1043-1052. DOI:10.1016/j.drudis.2017.11.009 |
| [12] |
S. Mura, J. Nicolas, P. Couvreur, Nat. Mater. 12 (2013) 991-1003. DOI:10.1038/nmat3776 |
| [13] |
M.R. Junttila, F.J. de Sauvage, Nature 501 (2013) 346-354. DOI:10.1038/nature12626 |
| [14] |
M. Wang, J. Zhao, L. Zhang, et al., J. Cancer 8 (2017) 761-773. DOI:10.7150/jca.17648 |
| [15] |
T. Wu, Y. Dai, Cancer Lett. 387 (2017) 61-68. DOI:10.1016/j.canlet.2016.01.043 |
| [16] |
M. Shahriari, M. Zahiri, K. Abnous, et al., J. Control. Release 308 (2019) 172-189. DOI:10.1016/j.jconrel.2019.07.004 |
| [17] |
S. Gao, G. Tang, D. Hua, et al., J. Mater. Chem. B 7 (2019) 709-729. DOI:10.1039/C8TB02491J |
| [18] |
Y. Tian, M. Lei, L. Yan, F. An, Polym. Chem. 11 (2020) 2360-2369. DOI:10.1039/D0PY00004C |
| [19] |
B. Renoux, F. Raes, T. Legigan, et al., Chem. Sci. 8 (2017) 3427-3433. DOI:10.1039/C7SC00472A |
| [20] |
H. Chen, Y. Jin, J. Wang, et al., Nanoscale 10 (2018) 20946-20962. DOI:10.1039/C8NR07146B |
| [21] |
D. Dheer, J. Nicolas, R. Shankar, Adv. Drug Deliv. Rev. 151-152 (2019) 130-151. DOI:10.1016/j.addr.2019.01.010 |
| [22] |
Y. Sheng, J. Hu, J. Shi, L. Lee, Curr. Med. Chem. 26 (2019) 2377-2388. DOI:10.2174/0929867324666170830102409 |
| [23] |
T. Hampton, JAMA 319 (2018) 2161-2162. DOI:10.1001/jama.2017.12861 |
| [24] |
Y. Li, L. Du, C. Wu, et al., Curr. Top. Med. Chem. 19 (2019) 74-97. DOI:10.2174/1568026619666190125144621 |
| [25] |
C.S. Gondi, J.S. Rao, Expert Opin. Ther. Targets 17 (2013) 281-291. DOI:10.1517/14728222.2013.740461 |
| [26] |
O.C. Olson, J.A. Joyce, Nat. Rev. Cancer 15 (2015) 712-729. DOI:10.1038/nrc4027 |
| [27] |
R. Henning, Biochim. Biophys. Acta 401 (1975) 307-316. DOI:10.1016/0005-2736(75)90314-4 |
| [28] |
R. Xing, A.K. Addington, R.W. Mason, Biochem. J. 332 (1998) 499-505. DOI:10.1042/bj3320499 |
| [29] |
T. Sun, D. Jiang, L. Zhang, et al., Oncol. Lett. 11 (2016) 575-583. DOI:10.3892/ol.2015.3960 |
| [30] |
V. Bellat, R. Ting, T.L. Southard, et al., Adv. Funct. Mater. 28 (2018) 1803969. DOI:10.1002/adfm.201803969 |
| [31] |
H. Kirschke, A.J. Barrett, N.D. Rawlings, Protein Profile 2 (1995) 1581-1643. |
| [32] |
I.M. Berquin, L. Cao, D. Fong, B.F. Sloane, Gene 159 (1995) 143-149. DOI:10.1016/0378-1119(95)00072-E |
| [33] |
J. Reiser, B. Adair, T. Reinheckel, J. Clin. Invest. 120 (2010) 3421-3431. DOI:10.1172/JCI42918 |
| [34] |
D. Musil, D. Zucic, D. Turk, et al., EMBO J. 10 (1991) 2321-2330. DOI:10.1002/j.1460-2075.1991.tb07771.x |
| [35] |
C. Illy, O. Quraishi, J. Wang, et al., J. Biol. Chem. 272 (1997) 1197-1202. DOI:10.1074/jbc.272.2.1197 |
| [36] |
J.C. Krupa, S. Hasnain, D.K. Nägler, et al., Biochem. J. 361 (2002) 613-619. DOI:10.1042/bj3610613 |
| [37] |
J. Schmitz, E. Gilberg, R. Löser, et al., Bioorg. Med. Chem. 27 (2019) 1-15. DOI:10.1016/j.bmc.2018.10.017 |
| [38] |
A. Mitrović, B. Mirković, I. Sosič, et al., Biol. Chem. 397 (2016) 1659-174. |
| [39] |
B. Mirković, B. Markelc, M. Butinar, et al., Oncotarget 6 (2015) 19027-19042. DOI:10.18632/oncotarget.3699 |
| [40] |
J. Kos, A. Mitrović, B. Mirković, Future Med. Chem. 6 (2014) 1355-1371. DOI:10.4155/fmc.14.73 |
| [41] |
J.S. Mort, D.J. Buttle, Int. J. Biochem. Cell Biol. 29 (1997) 715-720. DOI:10.1016/S1357-2725(96)00152-5 |
| [42] |
T. Zhang, Y. Maekawa, J. Hanba, et al., Immunology 100 (2000) 13-20. DOI:10.1046/j.1365-2567.2000.00000.x |
| [43] |
H. Büth, P. Luigi Buttigieg, R. Ostafe, et al., Eur. J. Cell Biol. 86 (2007) 747-761. DOI:10.1016/j.ejcb.2007.03.009 |
| [44] |
H. Büth, B. Wolters, B. Hartwig, et al., Eur. J. Cell Biol. 83 (2004) 781-795. DOI:10.1078/0171-9335-00428 |
| [45] |
U. Felbor, B. Kessler, W. Mothes, et al., Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 7883-7888. DOI:10.1073/pnas.112632299 |
| [46] |
O. Mijanovć, A. Branković, A.N. Panin, et al., Cancer Lett. 449 (2019) 207-214. DOI:10.1016/j.canlet.2019.02.035 |
| [47] |
B. Tong, B. Wan, Z. Wei, et al., Clin. Exp. Immunol. 177 (2014) 586-597. DOI:10.1111/cei.12357 |
| [48] |
A.A. Aghdassi, D.S. John, M. Sendler, et al., J. Biol. Chem. 293 (2018) 1018-1029. DOI:10.1074/jbc.M117.814772 |
| [49] |
S. Hartmann, R. Lopez Cruz, S. Alameh, et al., ACS Infect. Dis. 4 (2018) 1235-1245. DOI:10.1021/acsinfecdis.8b00053 |
| [50] |
H. Nakanishi, Neural Regeneration Res. 15 (2020) 25-29. DOI:10.4103/1673-5374.264444 |
| [51] |
A. Baici, K. Müntener, A. Willimann, R. Zwicky, Biol. Chem. 387 (2006) 1017-1021. |
| [52] |
J.R. Pungercar, D. Caglic, M. Sajid, et al., FEBS J. 276 (2009) 660-668. DOI:10.1111/j.1742-4658.2008.06815.x |
| [53] |
B. Breznik, A. Mitrović, T.L. T, J. Kos, Biochimie 166 (2019) 233-250. DOI:10.1016/j.biochi.2019.05.002 |
| [54] |
O. Meurette, P. Mehlen, Cancer Cell 34 (2018) 536-548. DOI:10.1016/j.ccell.2018.07.009 |
| [55] |
M.M. Mohamed, B.F. Sloane, Nat. Rev. Cancer 6 (2006) 764-775. DOI:10.1038/nrc1949 |
| [56] |
E. Guerra, A. Cimadamore, P. Simeone, et al., BMC Cancer 16 (2016) 649. DOI:10.1186/s12885-016-2713-3 |
| [57] |
G. Kostoulas, A. Lang, H. Nagase, A. Baici, FEBS Lett. 455 (1999) 286-290. DOI:10.1016/S0014-5793(99)00897-2 |
| [58] |
J.E. Koblinski, M. Ahram, B.F. Sloane, Clin. Chim. Acta 291 (2000) 113-135. DOI:10.1016/S0009-8981(99)00224-7 |
| [59] |
O. Vasiljeva, B. Turk, Biochimie 90 (2008) 380-386. DOI:10.1016/j.biochi.2007.10.004 |
| [60] |
J. Mu, J. Lin, P. Huang, X. Chen, Chem. Soc. Rev. 47 (2018) 5554-5573. DOI:10.1039/C7CS00663B |
| [61] |
E. Li, Y. Yang, G. Hao, et al., Nanotheranostics 2 (2018) 233-242. DOI:10.7150/ntno.25565 |
| [62] |
H.J. Cho, S. Lee, S.J. Park, et al., Colloid Surf. B:Biointerfaces 179 (2019) 9-16. DOI:10.1016/j.colsurfb.2019.03.047 |
| [63] |
J. Ding, J. Chen, L. Gao, et al., Nano Today 29 (2019) 100800. DOI:10.1016/j.nantod.2019.100800 |
| [64] |
T.M. Allen, P.R. Cullis, Adv. Drug Deliv. Rev. 65 (2013) 36-48. DOI:10.1016/j.addr.2012.09.037 |
| [65] |
K. Zhao, D. Li, C. Shi, et al., Curr. Drug Deliv. 13 (2016) 494-499. DOI:10.2174/156720181304160521004609 |
| [66] |
I.A. Isoglu, Y. Ozsoy, S.D. Isoglu, Curr. Top. Med. Chem. 17 (2017) 1469-1489. DOI:10.2174/1568026616666161222110600 |
| [67] |
A.S. Chauhan, Molecules 23 (2018) 938. DOI:10.3390/molecules23040938 |
| [68] |
Y. Jiang, S. Huo, J. Hardie, et al., Expert Opin. Drug Deliv. 13 (2016) 547-559. DOI:10.1517/17425247.2016.1134486 |
| [69] |
X. Ma, L. Gao, Y. Tang, M. Peng, Part. Part. Syst. Charact. 35 (2017) 1700326. |
| [70] |
C.T. Matea, T. Mocan, F. Tabaran, et al., Int. J. Nanomed. 12 (2017) 5421-5431. DOI:10.2147/IJN.S138624 |
| [71] |
S. Sengupta, R. Sasisekharan, Br. J. Cancer 96 (2007) 1315-1319. DOI:10.1038/sj.bjc.6603707 |
| [72] |
X. Zhou, Y. Hao, L. Yuan, et al., Chin. Chem. Lett. 29 (2018) 1713-1724. DOI:10.1016/j.cclet.2018.10.037 |
| [73] |
P. Li, Y. Chen, Y. Liu, Chin. Chem. Lett. 30 (2019) 1190-1197. DOI:10.1016/j.cclet.2019.03.035 |
| [74] |
L. Guo, H. Chen, N. He, Y. Deng, Chin. Chem. Lett. 29 (2018) 1829-1833. DOI:10.1016/j.cclet.2018.10.038 |
| [75] |
K. Wieland, G. Ramer, V.U. Weiss, et al., Nano Res. 12 (2019) 197-203. DOI:10.1007/s12274-018-2202-x |
| [76] |
X. Fu, J. Cai, X. Zhang, et al., Adv. Drug Deliv. Rev. 132 (2018) 169-187. DOI:10.1016/j.addr.2018.07.006 |
| [77] |
M. Ranjbar, A. Pardakhty, A. Amanatfard, A. Asadipour, Drug Des. Devil. Ther. 12 (2018) 2635-2643. DOI:10.2147/DDDT.S173324 |
| [78] |
M.T. Alula, P. Lemmens, L. Bo, et al., Anal. Chim. Acta 1073 (2019) 62-71. DOI:10.1016/j.aca.2019.04.061 |
| [79] |
H. Peng, Q. Huang, T. Wu, et al., Curr. Drug Deliv. 15 (2018) 278-285. DOI:10.2174/1567201814666170224144217 |
| [80] |
W. Yu, T. Sun, C. Qi, et al., ACS Appl. Mater. Interfaces 9 (2017) 3306-3317. DOI:10.1021/acsami.6b12325 |
| [81] |
T. Sun, Y. Zhu, C. Qi, et al., J. Mater. Chem.B 4 (2016) 3257-3268. DOI:10.1039/C5TB02632F |
| [82] |
H. Liu, J. Mei, Y. Xu, et al., Int. J. Nanomed. 14 (2019) 8739-8751. DOI:10.2147/IJN.S224044 |
| [83] |
E.S. Dolinina, E.V. Parfenyuk, Curr. Drug Deliv. 14 (2017) 734-740. |
| [84] |
R. Fang, C. Hu, B.T. Luk, et al., Nano Lett. 14 (2014) 2181-2188. DOI:10.1021/nl500618u |
| [85] |
L.C. Glangchai, M. Caldorera-Moore, L. Shi, K. Roy, J. Control. Release 125 (2008) 263-272. DOI:10.1016/j.jconrel.2007.10.021 |
| [86] |
V. Bellat, H.H. Lee, L. Vahdat, B. Law, Biomacromolecules 17 (2016) 2040-2049. DOI:10.1021/acs.biomac.6b00227 |
| [87] |
R. Löser, J. Pietzsch, Front. Chem. 3 (2015) 37. |
| [88] |
E. Gounaris, C.H. Tung, C. Restaino, et al., PLoS One 3 (2008) e2916. DOI:10.1371/journal.pone.0002916 |
| [89] |
V. Lyo, F. Cattaruzza, T.N. Kim, et al., Am. J. Physiol. Gastrointest. Liver Physiol. 303 (2012) G894-G903. DOI:10.1152/ajpgi.00073.2012 |
| [90] |
D.V. Hingorani, L.A. Montano, E.A. Randtke, et al., Contrast Media Mol. Imaging 11 (2016) 130-138. DOI:10.1002/cmmi.1672 |
| [91] |
M.E. Roth-Konforti, C.R. Bauer, D. Shabat, Angew. Chem. Int. Ed. 56 (2017) 15633-15638. DOI:10.1002/anie.201709347 |
| [92] |
L.S. Ryan, A.R. Lippert, Angew. Chem. Int. Ed. 57 (2018) 622-624. DOI:10.1002/anie.201711228 |
| [93] |
Q. Wang, L. Yu, R.C.H. Wong, P.C. Lo, Eur. J. Med. Chem. 179 (2019) 828-836. DOI:10.1016/j.ejmech.2019.06.082 |
| [94] |
H. Shen, J. Liu, J. Lei, H. Ju, Chem. Commun. 54 (2018) 9155-9158. DOI:10.1039/C8CC04621B |
| [95] |
X. Gao, J. Li, M. Luan, et al., Biosens. Bioelectron. 147 (2020) 111755. DOI:10.1016/j.bios.2019.111755 |
| [96] |
R. Zhang, J. Yang, D.C. Radford, et al., Macromol. Biosci. 17 (2017) 1600125. DOI:10.1002/mabi.201600125 |
| [97] |
E. Kisin-Finfer, S. Ferber, R. Blau, et al., Bioorg. Med. Chem. Lett. 24 (2014) 2453-2458. DOI:10.1016/j.bmcl.2014.04.022 |
| [98] |
H. Li, C.H. Lee, I. Shin, Org. Lett. 21 (2019) 4628-4631. DOI:10.1021/acs.orglett.9b01530 |
| [99] |
G. Saranya, M.M. Joseph, V. Karunakaran, et al., ACS Appl. Mater. Interfaces 10 (2018) 38807-38818. DOI:10.1021/acsami.8b15583 |
| [100] |
L. Sun, X. Li, X. Wei, et al., ACS Appl. Mater. Interfaces 8 (2016) 10499-10512. DOI:10.1021/acsami.6b00980 |
| [101] |
Y. Wang, Y. Dai, Q. Luo, et al., J. Biomed. Nanotechnol. 15 (2019) 1384-1400. DOI:10.1166/jbn.2019.2759 |
| [102] |
S. Tran, P.J. DeGiovanni, B. Piel, P. Rai, Clin. Transl. Med. 6 (2017) 44. |
| [103] |
Y. Cheng, G. Luo, J. Zhu, et al., ACS Appl. Mater. Interfaces 7 (2015) 9078-9087. DOI:10.1021/acsami.5b00752 |
| [104] |
Z. Wang, R. Zhang, T. Zhang, et al., ACS Appl. Mater. Interfaces 10 (2018) 41056-41069. DOI:10.1021/acsami.8b14001 |
| [105] |
S.P. Tarassoli, A.M. de Pinillos Bayona, H. Pye, et al., Nanotechnology 28 (2017) 055101. DOI:10.1088/1361-6528/28/5/055101 |
| [106] |
Y. Li, Q. Dong, Y. Cui, Cancer Biol. Med. 16 (2019) 415-434. |
| [107] |
C. Zhang, L. Liu, W. Qiu, et al., Small 14 (2018) 1703321. DOI:10.1002/smll.201703321 |
| [108] |
F. Wang, M. Wang, L. Zhao, Q. Li, Mater. Express 9 (2019) 1076-1081. DOI:10.1166/mex.2019.1602 |
| [109] |
X. Duan, X. Yang, C. Dai, et al., Mater. Express 9 (2019) 757-763. DOI:10.1166/mex.2019.1552 |
| [110] |
Y. Luo, X. Sun, L. Huang, et al., ACS Appl. Mater. Interfaces 11 (2019) 29490-29497. DOI:10.1021/acsami.9b07390 |
| [111] |
Z. Guo, L. Shi, H. Feng, et al., Chin. Chem. Lett. 31(2020), doi: http://dx.doi.org/10.1016/j.cclet.2020.03.066.
|
| [112] |
L. Bildstein, C. Dubernet, P. Couvreur, Adv. Drug Deliv. Rev. 63 (2011) 3-23. DOI:10.1016/j.addr.2010.12.005 |
| [113] |
V. Bala, S. Rao, B.J. Boyd, C.A. Prestidge, J. Control. Release 172 (2013) 48-61. DOI:10.1016/j.jconrel.2013.07.022 |
| [114] |
Q. Guo, H. Wang, Y. Zhao, et al., Polym. Chem. 4 (2013) 4584-4587. DOI:10.1039/c3py00742a |
| [115] |
Y. Chen, W. Zhang, Y. Huang, et al., Int. J. Pharm. 488 (2015) 44-58. DOI:10.1016/j.ijpharm.2015.04.048 |
| [116] |
X. Duan, J. Xiao, Q. Yin, et al., ACS Nano 7 (2013) 5858-5869. DOI:10.1021/nn4010796 |
| [117] |
L. Gao, Z. Yu, D. Meng, et al., Protein Pept. Lett. 22 (2015) 762-766. DOI:10.2174/0929866522666150622101944 |
| [118] |
Y. Dai, X. Ma, Y. Zhang, et al., Biomater. Sci. 6 (2018) 2976-2986. DOI:10.1039/C8BM00946E |
| [119] |
C. Zhang, D. Pan, J. Li, et al., Acta Biomater. 55 (2017) 153-162. DOI:10.1016/j.actbio.2017.02.047 |
| [120] |
Y. Zhong, L. Shao, Y. Li, Int. J. Oncol. 42 (2013) 373-383. DOI:10.3892/ijo.2012.1754 |
| [121] |
S.J. Lee, Y.I. Jeong, H.K. Park, et al., Int. J. Nanomed. 10 (2015) 5489-5503. |
| [122] |
Z. Peng, J. Kopecek, J. Am. Chem. Soc. 137 (2015) 67269-6729. |
| [123] |
X. Wei, Q. Luo, L. Sun, et al., ACS Appl. Mater. Interfaces 8 (2016) 11765-11778. DOI:10.1021/acsami.6b02006 |
| [124] |
Y. Ou, K. Chen, H. Cai, et al., Biomater. Sci. 6 (2018) 1177-1188. DOI:10.1039/C8BM00095F |
| [125] |
Y. Liang, S. Li, X. Wang, et al., J. Control. Release 275 (2018) 129-141. DOI:10.1016/j.jconrel.2018.01.033 |
| [126] |
L. Qiu, L. Zhao, C. Xing, Y. Zhan, Chin. Chem. Lett. 29 (2018) 301-304. DOI:10.1016/j.cclet.2017.09.048 |
| [127] |
J. Li, F. Liu, Q. Shao, et al., Adv. Health. Mater. 3 (2014) 1230-1239. DOI:10.1002/adhm.201300613 |
| [128] |
H. Zhang, X. Zhao, L. Chen, et al., Anal. Chem. 92 (2020) 1179-1188. DOI:10.1021/acs.analchem.9b04318 |
| [129] |
H. Han, Q. Jin, Y. Wang, et al., Chem. Commun. 51 (2015) 17435-17438. DOI:10.1039/C5CC06654A |
| [130] |
H. Han, J. Wang, T. Chen, et al., J. Colloid Interface Sci. 507 (2017) 217-224. DOI:10.1016/j.jcis.2017.07.047 |
| [131] |
M.L. Yap, J.D. McFadyen, X. Wang, et al., Theranostics 9 (2019) 1154-1169. DOI:10.7150/thno.29146 |
| [132] |
L. Huang, Y. Luo, X. Sun, et al., Biosens. Bioelectron. 92 (2017) 724-732. DOI:10.1016/j.bios.2016.10.004 |
| [133] |
Y. Yang, Q. Chen, S. Li, et al., J. Biomed. Nanotechnol. 14 (2018) 1396-1408. DOI:10.1166/jbn.2018.2592 |
| [134] |
X. Li, Y. Zhang, Z. Ma, et al., Chin. Chem. Lett. 30 (2019) 489-493. DOI:10.1016/j.cclet.2018.03.019 |
| [135] |
J. Tian, L. Ding, Q. Wang, et al., Anal. Chem. 87 (2015) 3841-3848. DOI:10.1021/acs.analchem.5b00429 |
| [136] |
Y.I. Jeong, S.Y. Yoo, J. Heo, D.H. Kang, ACS Omega 4 (2019) 18593-18599. DOI:10.1021/acsomega.9b02386 |
| [137] |
J. Kim, C.H. Tung, Y. Choi, Chem. Commun. 50 (2014) 10600-10603. DOI:10.1039/C4CC04166F |
| [138] |
X. Chen, D. Lee, S. Yu, et al., Biomaterials 122 (2017) 130-140. DOI:10.1016/j.biomaterials.2017.01.020 |
| [139] |
J. Liu, L. Zhang, J. Lei, et al., ACS Appl. Mater. Interfaces 9 (2017) 2150-2158. DOI:10.1021/acsami.6b14446 |
| [140] |
Y. Yang, J. Aw, K. Chen, et al., Chem. Asian J. 6 (2011) 1381-1389. DOI:10.1002/asia.201000905 |
| [141] |
G. Mikhaylov, D. Klimpel, N. Schaschke, et al., Angew. Chem. Int. Ed. 53 (2014) 10077-10081. DOI:10.1002/anie.201402305 |
| [142] |
F. An, Z. Yang, M. Zheng, et al., J. Nanobiotechnol. 18 (2020) 49. DOI:10.1186/s12951-020-00603-8 |
| [143] |
J. Li, Z. Tian, X. Ge, et al., Eur. J. Med. Chem. 163 (2019) 830-839. DOI:10.1016/j.ejmech.2018.12.021 |
| [144] |
S. Ferber, H. Baabur-Cohen, R. Blau, et al., Cancer Lett. 352 (2014) 81-89. DOI:10.1016/j.canlet.2014.02.022 |
| [145] |
S.M. Shon, Y. Choi, J.Y. Kim, et al., Arterioscler. Thromb. 33 (2013) 1360-1365. DOI:10.1161/ATVBAHA.113.301290 |
| [146] |
B. Wang, P. Lv, H. Cai, et al., J. Biomed. Nanotechnol. 15 (2019) 1897-1908. DOI:10.1166/jbn.2019.2833 |
| [147] |
H. Gao, H. Feng, Y. Bai, et al., J. Biomed. Nanotechnol. 15 (2019) 1764-1770. DOI:10.1166/jbn.2019.2805 |
| [148] |
S.T. Gunawan, K. Liang, G.K. Such, et al., Small 10 (2014) 4080-4086. |
| [149] |
M. Fathi, A. Safary, J. Barar, Bioimpacts 10 (2020) 1-4. |
| [150] |
C. Hu, A. Qian, Q. Wang, et al., Eur. J. Pharm. Biopharm. 109 (2016) 206-213. DOI:10.1016/j.ejpb.2016.10.018 |
| [151] |
H. Jia, Y. Zhu, Q. Duan, et al., J. Control. Release 311-312 (2019) 301-318. DOI:10.1016/j.jconrel.2019.08.022 |