 2020, Vol. 31
2020, Vol. 31 


b University of Science and Technology of China, Hefei 230026, China;
c Jilin Biomedical Polymers Engineering Laboratory, Changchun 130022, China
Polypeptides, as one kind of biocompatible and biodegradable polymers, have attracted the attention of scientists as promising biomaterials. A wide range of polypeptides composed of various α-amino acid residues as structural units possess several potential advantages listed below for biomedical applications [1]. (i) Diversity of the chemical structure [2]. Different polypeptides can be synthesized by the ring-opening polymerization (ROP) of various α-amino acid N-carboxyanhydrides (NCAs), and modified with functional groups and bioactive agents to meet the practical requirements. (ii) Different scales of devices [3]. Through tuning the compositions, sequences, and polymerization degrees of polypeptides, they are constructed into the nanoscale, microscale, and macroscale devices by different techniques. (iii) Excellent biocompatibility and biodegradability [4]. Polypeptides have favorable biocompatibility as a benefit of the α-amino acid components and controlled biodegradation in the presence of specific enzymes in vivo. Thus, a wide range of polypeptides and their composites have been tailored for biological, medical, and pharmaceutical applications, such as controlled drug release, targeted gene delivery, theranostics, and even regenerative medicine [3, 5-8].
For the successful clinical applications, the precise chemical structure and low polydispersity index (PDI) are critical factors. Over the past several decades, massive efforts have been made to synthesize polypeptides with predictable molecular weights, narrow PDIs, precision peptide sequences, and controlled physicochemical properties. Generally, there are mainly three chemical methods for polypeptide synthesis, including solid phase peptide synthesis (SPPS), solution phase coupling, and ROP of α-amino acid NCAs [9]. SPPS is a routine technique that is able to obtain polypeptides whose sequences can be precisely controlled by the successive coupling of α-amino acids. While some problems, such as the limited achievable maximum chain length (only to about 50 α-amino acid residues), low yield, poor economy, and possible racemization, restrict its development and broad application [10, 11]. Instead of α-amino acids, solution-coupling methods utilize short peptide sequences as starting materials to prepare more abundant polymers via a step-growth process. Thus, this kind of method is always employed for the fabrication of polypeptides containing repeating short sequences (ca. 3–10 residues in length). However, this technique is plagued with the difficulty of synthesizing starting oligopeptides, cyclization reactions, and low molecular weights of products [9].
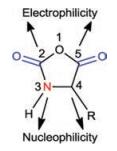
The ROP of α-amino acid NCAs is the most widely accepted strategy for the facile preparation of polypeptides with high molecular weights [12]. The chemical structure of α-amino acid NCAs is shown in Fig. 1, in which the carbon atoms of 2-carbonyl (2-CO) and 5-carbonyl (5-CO) groups are electrophilic while the 3-nitrogen atom and 4-carbon atom are nucleophilic. Leuchs and Hermann first reported the successful preparation and polymerization of α-amino acid NCAs in 1906 [13-16]. After 1921, Curtius et al. used water, alcohol, or primary amines to initiate ROP of α-amino acid NCAs to prepare high molecular weight polypeptides [17-24].

|
Download:
|
| Fig. 1. Structure and reactive centers in α-amino acid NCA. | |
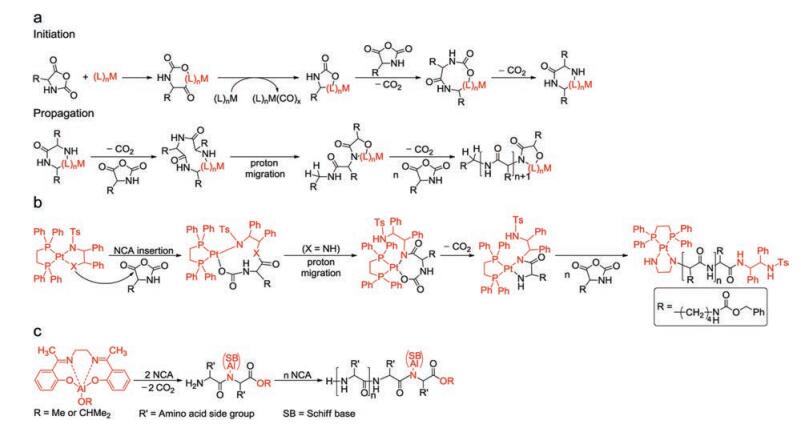
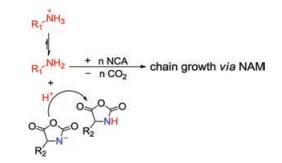
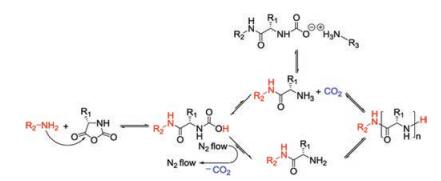
There are generally two recognized pathways when nucleophiles or bases initiate the ROP of α-amino acid NCAs, which are "normal amine mechanisms" (NAM) and the "active monomer mechanisms" (AMM). The NAM is usually adopted when the ROP of α-amino acid NCAs is initiated by nonionic initiators possessing one or more mobile hydrogen atoms, such as primary amines, alcohols, and thiols. The initiation reaction proceeds via the nucleophilic attack on the electron-deficient carbonyl carbon atom (2-CO) of the NCA ring [25]. The ring opens, and the unstable intermediate carbamic acid forms by proton transfer, which subsequently decarboxylates by the release of carbon dioxide (CO2) to obtain a free amino group as a new attacker to maintain the polymerization (Fig. 2a). This mechanism allows for preparing products with controllable molecular weights and predictable end-groups. Because of existing trace impurities in α-amino acid NCAs or solvent, or the presence of water, however, various possible side reactions, such as the disablement of the active end groups or the formation of cyclic structures, would occur and lead to reduced reaction efficacy [25-30].

|
Download:
|
| Fig. 2. Mechanisms for ROP of α-amino acid NCAs. (a) Normal amine mechanism. (b) Active monomer mechanism. Reproduced with permission [9]. Copyright 1997, John Wiley & Sons, Inc. | |
The AMM is depicted as Fig. 2b. It would be the prior pathway when bases (i.e., tertiary amines) or alkoxides that extract the proton from the nitrogen (4-N) of α-amino acid NCAs exist so that they lead to the formation of NCA anions. Then this anion attacks the 2-CO of another NCA to give a tadpole dimer, followed by reaction with a third NCA, creating a new anion after the elimination of CO2. By analogy, the new anion is able to attack the fore-mentioned tadpole dimer to promote chain propagation. Thus, to be precise, bases act as catalysts rather than chain initiators in this procedure. The two standards of living polymerization are the retention of reactive end groups in the polymer chains and low PDIs. Only when the initiation step is faster than the growing step can a low PDI be achieved. So sterically unhindered primary aliphatic amines are prior to tertiary amines in terms of selecting initiators. Moreover, the AMM adopted when tertiary amines are utilized as initiators is generally entirely uncontrolled and thus less favored in the synthesis of well-defined polypeptide compounds. These two pathways tend to coexist during polymerization reactions, which brings about difficulties in synthesizing polypeptides with high molecular weights, predictable structures, and varied architectures [9].
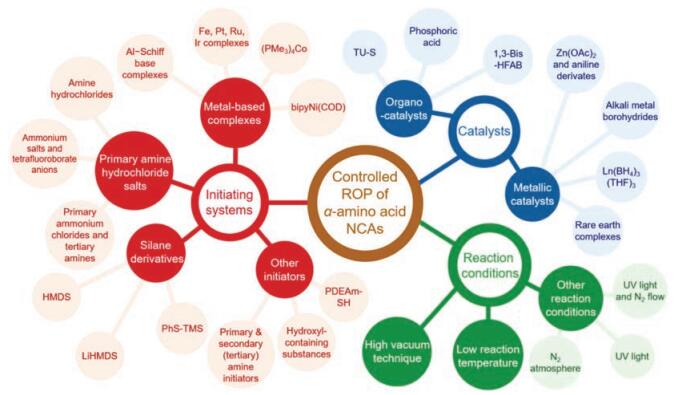
Controlled synthesis of polypeptides through the ROP of α-amino acid NCAs has appealed the attention of researchers for decades. Since the first living initiating system published by Deming in 1997, various efficient living initiating systems have been studied [31]. Apart from these, the reaction conditions have been optimized, ranging from pressure, temperature, light, to nitrogen atmosphere. In addition, several catalysts have been developed, including metallic and organic catalysts. They together make the preparation of polypeptides with controllable molecular weights, low PDIs, sophisticated polymeric architectures, and satisfying optical purities in high yield come true [12, 32]. In this review, we will give an overview of up-to-date improvements in techniques for the controlled ROP of α-amino acid NCAs through three aspects above, as shown in Scheme 1. These approaches provide a large scale of polypeptides for various practical applications [33].

|
Download:
|
| Scheme 1. Strategies for controlled ROP of α-amino acid NCAs. | |
2. Controlled synthesis of polypeptides through selection of initiating systems 2.1. Metal-based complexes
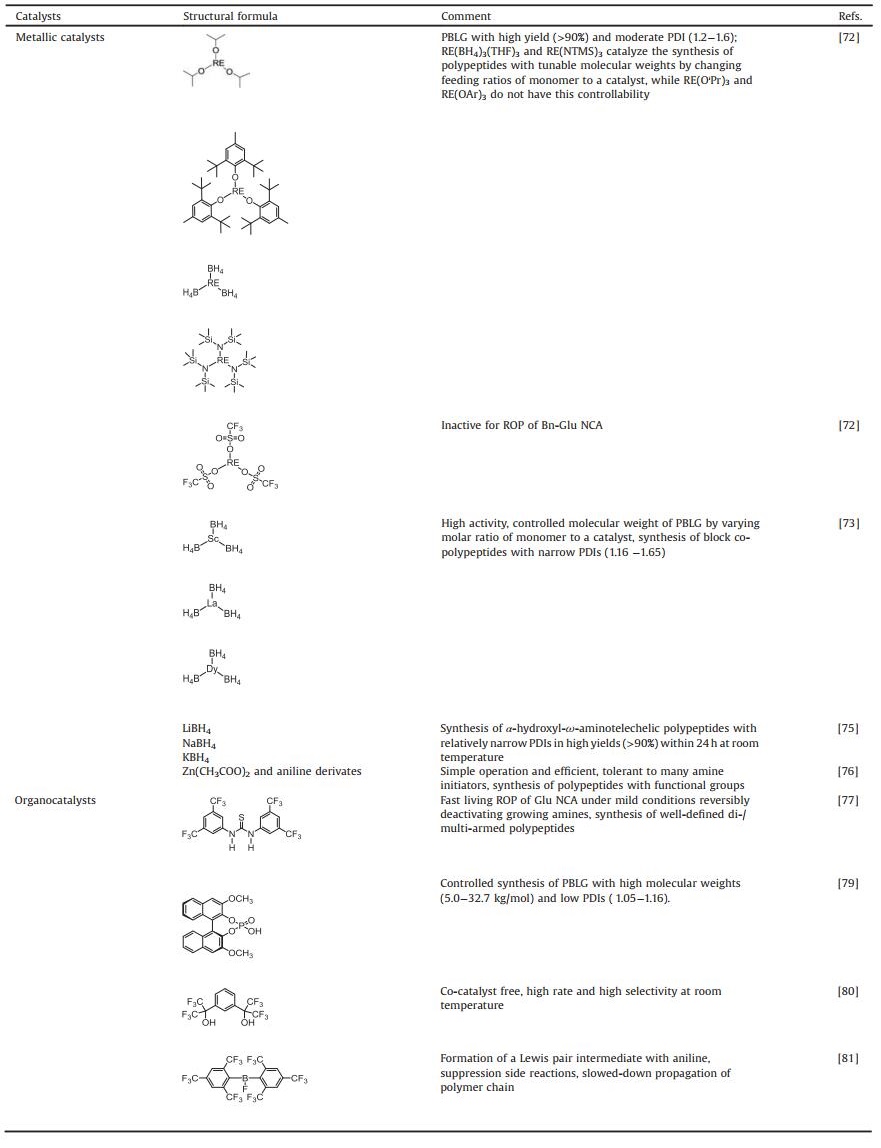
The synthesis of well-defined polypeptides is impeded by unwanted side reactions, e.g., chain-transfer and termination, that give rise to products with large PDIs [13]. In order to diminish side reactions, various initiating systems have been explored to obtain products similar to theoretical ones. The structural formulas and corresponding features of different initiators are presented in Table 1 in the order that they are mentioned. To begin with, metallic compounds have been exploited as initiators for ROP so that the side reactions can be suppressed. Among them, it was the discovery of organonickel (Ni) complexes, namely bipyNi(COD) (bipy = 2, 20-bipyridyl, COD = 1, 5-cyclooctadiene), by Deming in 1997 that the synthesis of high molecular weight homo-/co-polypeptides with low PDIs was achievable [31]. They were able to overcome possible termination reactions to realize predictable products.
|
|
Table 1 Initiating systems of controlled polymerization of α-amino acid NCAs. |

After that, Deming reported that cobalt (Co) initiators, e.g. (PMe3)4Co, were also a kind of promising candidates [34]. The forming of chelating metallacyclic intermediates is necessary via the oxidative addition of the transition metal complexes to the NCA rings. The intermediate reacts with a second NCA monomer to obtain a 6-membered amido-alkyl metallacycle, which then reacts with another NCA, accompanied by CO2 releasing and proton migration, and ultimately produces an active 5-membered amido-amidate complex to continue the polymerization reaction. The nucleophilic amido group of 5-membered amido-amidate complex then attacks in the electro-deficient 2-CO of new NCA to regenerat an active species, which is similar to the reaction initiated by bipyNi (COD), as shown in Fig. 3a. The terminal metal residues of the polymer chains can be eliminated efficiently from the polymers by precipitation or dialysis. The initiators based on Co and Ni can generate well-defined polypeptides with controlled molecular weights (500 < Mn < 500, 000 Da), low PDIs (< 1.2), and sophisticated stereoregularities [35]. Given that the initiating centers are generated by in situ reactions between metallic initiators and α-amino acid NCAs, the structures of polymer chain ends depend on the first added NCA. So it is difficult to obtain products with functionalized chain ends. To deal with this problem, Deming et al. developed amido-amidate metallacycle initiators containing nickel (Ni), which were utilized to synthesize polypeptides with functionalized chain ends, such as allyl and poly(polyethylene glycol) (PEG) [36].

|
Download:
|
| Fig. 3. Mechanisms for ROP of α-amino acid NCAs initiated by (a) bipyNi(COD) or (PMe3)4Co complexes. Reproduced with permission [34]. Copyright 1999, American Chemical Society. (b) Pt complexes. Reproduced with permission [37]. Copyright 2008, American Chemical Society. (c) Al–Schiff base complexes. Reproduced with permission [40]. Copyright 2003, American Chemical Society. | |
Besides Co and Ni-based compounds, iron (Fe) [34], platinum (Pt) [37, 38], rhodium (Ru) [39], iridium (Ir) [39], and aluminum (Al) complexes [40] have also been reported as active initiators for the ROP of α-amino acid NCAs. For example, Deming and co-workers have reported two Ru and Ir complexes coordinated by amido-sulfonamidate ligand. These complexes have been illustrated as potent initiators for the living ROP of α-amino acid NCAs [39]. Particularly, they provided a means to realize stereocontrol in NCA polymerization, which had been difficult to achieve in other metallacycle-based systems. In 2008, Liu et al. successfully discovered platinum complex ((dppe)Pt(MBS-NH), dppe = bis(diphenylphosphino)ethane, MBSNH = N-((1S, 2R)-2-amido-1, 2-diphenylethyl)-4-methylbenzene-sulfonamidato), which was an efficient initiator for the living ROP of N(ε)-benzyloxycarbonyl-L-lysine NCA (Cbz-Lys NCA) [37]. In the meanwhile, they proposed a mechanism for the ROP reaction, which was shown in Fig. 3b. In addition, a kind of novel Al–Schiff base complexes was reported by Bhaw-Luximon et al. to initiate the polymerization of γ-methyl glutamate NCA (Me-Glu NCA) at ambient temperature [40]. As shown in Fig. 3c, it is demonstrated that the polymerization is accompanied by the insertion of an NCA ring into the Al-alkoxy bond and subsequent loss of CO2 to generate the ester group at the chain ends. Then the terminal amino group initiates the ring-opening of the next NCA, obtaining an initiating amino group that allows the polymerization proceeding following the NAM pathway.
2.2. Primary amine hydrochloride saltsAs mentioned above, NAM and AMM tend to concurrent during ROP when the primary amine is employed as initiator. The AMM, however, suffers from the problem that it produces polypeptides plagued with relatively high PDIs because of a faster propagation rate compared with slow initiation. So it is desirable to come up with an effective strategy to eliminate, or at least limit, the AMM pathway during polymerization to realize living polymerization. Several researches have been reported to suppress the AMM pathway via improvement for existing strategies.
One of the prominent strategies is the use of amine hydrochlorides put forward by Dimitrov et al., which realized living ROP through preventing the protonation of prime amine groups from transforming into hydrochloric salts [41]. In acidic conditions, the formation of unwanted NCA anions was suppressed to a large extent, and meanwhile, CO2 was eliminated from reactive intermediates, which synergistically promoted the propagation. Moreover, thanks to the reversible formation of terminal amine hydrochlorides, most terminal amino groups existed as hydrochlorides salts, which impeded the AMM largely and prevented the happening of chain-termination. This mechanism was similar to living radical polymerization, which relied on the reversible activation and inactivation of initiating centers (Fig. 4). The obtained polypeptides, characterized with remarkably narrow PDIs (< 1.03), however, suffered from low conversion rate (70%–80%) due to low reactivity of amine hydrochloride even under high temperatures (40–80 ℃). Generally speaking, this is a promising living initiating system to generate well-defined products through further optimization.

|
Download:
|
| Fig. 4. Ammonium-mediated ROP of α-amino acid NCA. Reproduced with permission [41]. Copyright 2003, Royal Society of Chemistry. | |
The living polymerization of α-amino acid NCAs realized with metal-based initiating systems may bring in toxic substances indispensably. So extra postprocessing of the as-synthesized polymer, such as dialysis, is usually required, which might be costly and tedious. Especially in respect of the application in the whole field of nanomedicine, it is significant to develop methods that can produce a large-scale (from 100 g to kg) polypeptides without any trace of toxic impurities conveniently. In 2013, Conejos-Sánchez et al. devised initiators combining ammonium salts and non-nucleophilic tetrafluoroborate anions. They facilitated the preparation of a large number of polyglutamate with defined molecular weights (up to 800 units), low PDIs (< 1.2), controlled chain-end functionality, and adequate stereoselectivity [42]. The deprotection protocol in these methods enabled acidic as well as basic deprotection of poly(γ-benzyl-L-glutamate) (PBLG), which allowed the synthesis of PEG-based block copolymers and the incorporation of sensitive functional groups. The strategy can be widely applied to synthesize other polypeptides or polypeptide constructs.
More recently, a unique versatile way for controlled ROP of α-amino acid NCAs by using initiating systems composed of primary ammonium chlorides and tertiary amines was published by Vacogne et al. [43]. The rate of polymerization could be controlled by varying the ratio of primary ammonium salts to tertiary amines, and the reaction could even be paused and resumed by the addition of hydrochloric acid or triethylamine, respectively. Interestingly, despite proceeding through competing chain growth mechanisms, NAM and AMM, the methods afforded polypeptides with defined end groups molar masses, as well as low PDIs.
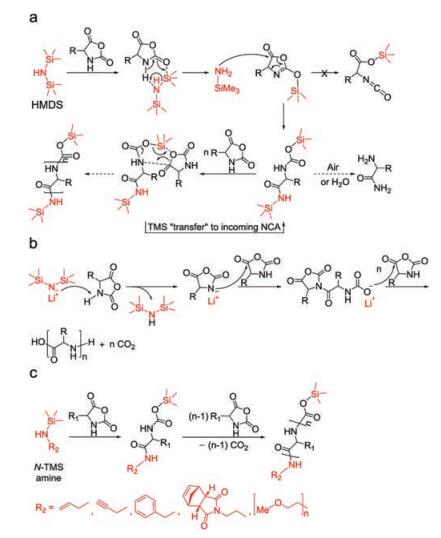
2.3. Silane derivativesDiffering from NAM and AMM, a polymerization pathway was presented in 2007 based on research conducted by Cheng and his colleagues who found that hexamethyldisilazane (HMDS) showed remarkable control over polymerizations of γ-benzyl-L-glutamate NCA (Bn-Glu NCA) to generate PBLG with expected molecular weights and narrow PDIs [44]. The molecular weights of PBLG showed a linear correlation with the conversion of Bn-Glu NCA and was consistent with the expected ones, demonstrating that PBLG chains propagated through the living chain ends. In the initiation step, accompanied by the cleavage of N-Si bond, the HMDS deprotonated the 3-N to form trimethylsilyl amine that attacked the 5-C to obtain a trimethylsilyl carbamate (TMS-CBM). TMS-CBM acted as the reactive active center, which could transfer the TMS group to the α-amino acid NCA to form a terminal TMS-CBM group (Fig. 5a). Interestingly, this process is similar to the group transfer polymerization of acrylic monomers initiated by organosilicon compounds. Therefore, HMDS is a potent reagent that allows a metal-free pathway for the controlled preparation of homo-/block-polypeptides with predictable molecular weights and low PDIs.

|
Download:
|
| Fig. 5. Proposed mechanisms for ROP of α-amino acid NCAs initiated by silane derivatives. (a) HMDS-mediated NCA polymerization through TMS-CBM group. Reproduced with permission [44]. Copyright 2007, American Chemical Society. (b) LiHMDS-initiated NCA polymerization. Reproduced with permission [45]. Copyright 2018, Springer Nature. (c) N-TMS amines as initiators to introduce functional group in polypeptides. Reproduced with permission [46]. Copyright 2008, American Chemical Society. | |
Based on this pioneering work, Wu et al. reported that lithium hexamethyldisilazide (LiHMDS) initiated an extremely rapid NCA polymerization process that completed within minutes or hours and could be conducted in an open vessel [45]. Polypeptides with variable chain lengths (degree of polymerization (DP) = 20–1294) and narrow molecular weight distributions (PDIs = 1.08–1.28) were readily prepared with this approach. Mechanism studies supported an anionic ROP mechanism, as presented in Fig. 5b. Conventional NCA polymerization strategies were plagued with high sensitivity to moisture, while in sharp contrast, LiHMDS-initiated NCA polymerization could be operated in an open flask outside of the glovebox without protection. This method enables quick library preparation and large-scale synthesis of polypeptides.
Given that the chain propagation relies on intermediate TMS-CBM and has nothing to do with the other TMS connected with nitrogen atom of HMDS, it is likely feasible to replace it with other functional units to offer various properties for polypeptides, which is what Cheng's group do afterward. They designed a series of N-TMS amines, including benzylamine, morpholine, propargylamine [46], N-(amino ethylene)-5-norbornene-endo-2, 3-dicarboximide [47], and mPEG2000 amine, which were all proved to be efficient initiators, as listed in Fig. 5c. The contained functional groups allowed polypeptides to be modified further through reactions, such as "click" chemistry and ring-opening metathesis polymerization.
More recently, Yuan et al. proposed the first example of trimethylsilyl sulfide (S-TMS), namely phenyl trimethylsilyl sulfide (PhS-TMS), mediating controlled the ROP of α-amino acid NCAs [48]. PhS-TMS could mediate rapid ROP of a broad scope of NCA monomers, producing functional polypeptides with an in situ generated phenyl thioester group at the carboxyl terminal. Compared with previously reported HMDS initiators, it offered more rapid chain initiation, which ensured a living polymerization with better control. This method constructs a powerful platform enabling the rapid generation of functional polypeptides and their carboxyl-terminal conjugates for numerous biological applications [49, 50]. Similarly, based on previous work, Lu's group reported an initiation system in 2018 based on a sulfur-stannum Lewis pair, trimethylstannyl phenylsulfide(PhS-SnMe3) [51].This initiator yielded polypeptides with high molecular weights (> 1.0×105 g/mol) and low PDIs within a few hours.
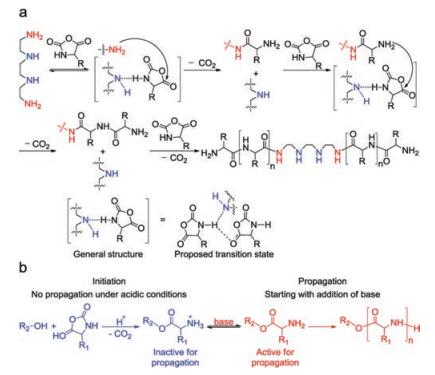
2.4. Other initiatorsBesides above mentioned three kinds of initiating systems, there are other compounds, which also can facilitate the living ROP of α-amino acid NCAs. For example, differing from conventional primary amine-mediated polymerizations only, a highly efficient strategy, based on an "alliance" of primary and secondary amine initiators, was successfully devised by Zhao et al. in 2015 [52]. It allowed the fast and living polymerization of α-amino acid NCAs at room temperature to generate well-defined products through the pathway of the so-called accelerated amine mechanism through monomer activation (AAMMA) (Fig. 6a). Subsequently, they further investigated the potential of initiators comprising of both primary and tertiary amines [53]. Interestingly, rather than proceeding fast and uncontrollably due to the assumed concurrence of AMM and NAM, actually it was observed that triethyla-minetriamine, a compound that contained both a sterically hindered tertiary amine and three primary amines, did bring about a fast and yet perfectly controlled ROP of α-amino acid NCAs.

|
Download:
|
| Fig. 6. Other mechanisms for ROP of α-amino acid NCAs initiated by (a) the "alliance" of primary and secondary amine initiators. Reproduced with permission [52]. Copyright 2015, American Chemical Society. (b) hydroxyl-containing substances. Reproduced with permission [54]. Copyright 2017, American Chemical Society. | |
As a kind of easily available reagents, the hydroxyl-containing substances are able to initiate the ROP of α-amino acid NCAs, while slow initiation rate enormously hinders their practical applications. Thanks to the researches of David's group in 2017, well-defined polypeptides initiated by the hydroxyl groups were prepared by taking advantage of acidic catalysts and subsequent addition of bases [54]. In this way, slow initiation was separated from the fast chain propagation step. After the completion of the initiation in an acidic environment, the propagation would start after the addition of bases to deprotonate the ammonium groups, as presented in Fig. 6b. This method was successfully applied for the synthesis of homopolypeptides as well as polypeptide-based block copolymers.
Similarly, thiols can also function as initiators for the ROP of α-amino acid NCAs. Zhang et al. [55] proposed a novel synthetic pathway for block polypeptides employing thiol-terminated macroinitiators instead of convention amino-terminated ones combining the ROP and RAFT reaction. The thiol-terminated poly-(N, N-diethylacrymide) (PDEAm-SH) macroinitiator was obtained via the selective aminolysis of a dithioester moiety resulting in the RAFT polymerization of N, N-diethylacrymide. The propagation of α-amino acid NCA initiated by a thiol group seemed to follow NAM, as shown during the homopolymerization of Bn-Glu NCA and N(ε)-trifluoroacetyl-lysine NCA (TFA-Lys NCA).
3. Controlled synthesis of polypeptides through optimization of reaction conditions 3.1. High vacuum techniquePredictable polypeptides can be obtained if the ROP proceeds only following the NAM pathway. However, NAM is susceptible to a range of impurities, such as hydrochloride and acyl chlorides (coming from the synthesis of NCAs), leading to termination reaction, as well as other initiating species (water and amines) resulting in wide PDIs. In addition, the presence of CO2 retards the reaction progress and leads to incomplete polymerization. To deal with these problems, a highly effective strategy for living polymerization of NCAs, high vacuum techniques (HVT), was proposed by Aliferis and co-workers in 2004 [56]. It created and maintained conditions necessary for the preparation of well-defined homo-/block-polypeptides with controllable chain lengths, low PDIs, and various architectures. The satisfactory products could be attributed to two following reasons. Firstly, HVT ensured that all reagents, and also the reaction environments, were completely impurity-free in all steps of the synthesis. Thus, side reactions induced by trace existence of water and other reactive impurities could be dramatically suppressed. Secondly, CO2 produced during the reaction was removed continually under highvacuum conditions so that the decomposition of carbamic acid was promoted to accelerate the chain propagation effectively via the NAM. After that, the star-shaped or more elaborated macromolecular structures were conducted successfully via HVT [57, 58].
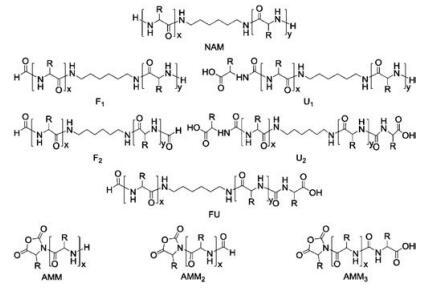
The reason why propagation performed more effectively under a high vacuum than normal pressure was illustrated systematically by Pickel et al. [59]. They demonstrated that the high vacuum polymerization of O-benzyl-L-tyrosine NCA (Bn-Tyr NCA) initiated by 1, 6-diamino hexane (DAH) proceeded exclusively through NAM with trace termination product F2, as listed in Fig. 7. In contrast, chain termination products of traditional polymerization were more complex than those produced under HVT, including AMM2, AMM3, F1, F2, and U2, because it proceeded via NAM and AMM concurrently.

|
Download:
|
| Fig. 7. Possible termination products in polymerization of Bn-Tyr NCA initiated by DAH. Reproduced with permission [59]. Copyright 2009, American Chemical Society. | |
Compared with the HVT mentioned above, the reduction of polymerization temperature is more direct and operable. It does not require the use of specific catalysts or demanding reaction conditions like HVT needed. An innovative strategy, namely low reaction temperature, which could eliminate side reactions during the NAM polymerization, was reported by Vayaboury et al. in 2004 [60]. At room temperature, the n-hexylamine-initiated polymerization of TFA-Lys NCA in N, N-dimethylformamide (DMF) produced polypeptides with broad molecular weight distributions because of the existence of "dead chains" generated by side reactions. It was quantified that there were 78% of dead polymers after the completion of polymerization at room temperature in this research. While at 0 ℃, there was almost no "dead polymer", accurately 1% "dead polymer" formed, testifying that possible living polymerization proceeded at low temperatures with intensive suppression of termination reactions. The living propagation could be attributed to higher activation energies of the side reactions than that of chain propagation at low temperatures [61]. What must be pointed out is that, however, the polymerization at 0 ℃ needs long reaction times owning to slow kinetics.
In addition, Habraken et al. investigated the homo, block, and graft polymers of various α-amino acid NCAs at 0 ℃ to verify the broad applicability of low-temperature techniques (Fig. 8) [62]. It was confirmed that frequently occurring end-group termination reactions between amino groups and solvent DMF or intramolecular side chains were nearly absent at 0 C. The polymerization was thus controlled, and homo-/co-polypeptides with low PDIs, around 1.1, were obtained.

|
Download:
|
| Fig. 8. List of α-amino acid NCAs applied in polymerization at 0 ℃. Reproduced with permission [62]. Copyright 2010, Royal Society of Chemistry. | |
In consideration of the high efficacy of HVT, they further studied polymerizations of a range of NCAs to optimize the reactions combining the high-vacuum and low-temperature techniques [63]. The synergistic effect of two different conditions not only maintained the living NCA polymerization but also solved the problem of long reaction time encountered in low-temperature technique. For example, NCA monomers of Bn-Glu, Cbz-Lys, and L-alanine (Ala) had considerably short polymerization time when a low pressure of 1 × 10-5 bar was applied. Meanwhile, it is detected that the formation of side products mostly started after full monomer conversion. In contrast, the NCA monomers of β-benzyl-L-aspartate (Bn-Asp), O-benzyl-L-serine (Bn-Ser), and O-benzyl-L-threonine (Bn-Thr) polymerized slowly even at low pressure. Furthermore, a tetrablock copolymer with low PDI (1.3) and high structural control comprising PBLG, poly(L-alanine), poly(N (ε)-benzyloxycarbonyl-L-lysine), and poly(β-benzyl L-aspartate) blocks were prepared via the combination of the optimal parameters of pressure and tempertaure.
In 2012, Cao et al. synthesized PBLG initiated by n-hexylamine in DMF under normal pressure at 0 ℃ [64]. It was observed that over 90% of the products were amino-terminated PBLG with low PDI when the polymerization was carried out at 0 ℃, which thus could be utilized to re-initiate the polymerization of other NCAs. Then several well-defined PBLG-containing block co-polypeptides were successfully synthesized conveniently.
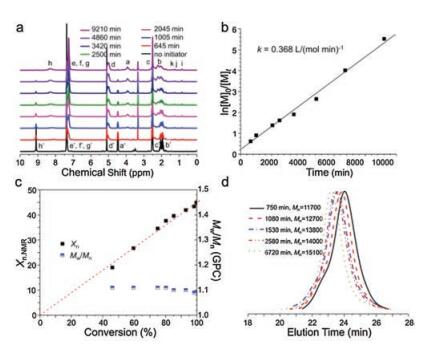
More recently, polymerization of Bn-Glu NCA was performed under frozen condition (cryo-ROP) for the first time by He's group [65]. This cryo-ROP proceeded in dimethyl sulfoxide (DMSO) or 1, 4-dioxane below the freezing point of polymerization mixture, which offset the slow polymerization rate in low-temperature conditions. It presented the excellent first-order kinetics and good control over molecular weight and molecular distribution (Fig. 9).

|
Download:
|
| Fig. 9. Kinetics and living properties of cryo-ROP. (a) Variation of 1H NMR spectrum of cryo-ROP mixture with time; (b) Linear dependence of ln([M]0/[M]t) on t; (c) Proportional dependence of Xn on conversion and the related PDI (Mw/Mn); (d) Typical GPC traces of PBLG at different polymerization periods (DMSO-d6, -18 ℃, [Bn-Glu NCA]0 = 0.0767 mol/mL, [Bn-Glu NCA]0/[butylamine]0 = 50). Reproduced with permission [65]. Copyright 2018, Elsevier Ltd. | |
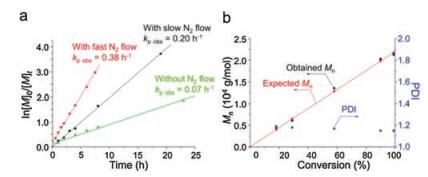
As known to all, the immediate removal of CO2 produced during reactions affects the kinetics of NCA polymerizations [66]. In contrast, the relationship between "livingness" of the ROP of NCAs and CO2 elimination had not been demonstrated all the time. Fortunately, in 2013, Zou et al. reinvestigated the influence of CO2 removal, confirming the relationship between nitrogen (N2) atmosphere and the living characteristics for the ROP of NCAs utilizing Bn-Glu NCA as a monomer and n-hexylamine as an initiator (Fig. 10) [67]. N2 flow method, as an easily-operational method, offered an approach for the synthesis of polypeptides with accelerated polymerization processes, predictable molecular weights, and low PDIs. This approach is attractive not only because most of the initiators used are commercially available, but there is no need to remove catalysts after polymerization. The N2 flow could be easily tuned to balance the polymerization kinetics and structural control in the ROP of NCAs, as shown in Fig. 11. The significantly accelerated polymerization decreased side reactions that might be introduced by moisture and solvent [68].

|
Download:
|
| Fig. 10. ROP of α-amino acid NCA under N2 flow via NAM. Reproduced with permission [67]. Copyright 2013, American Chemical Society. | |

|
Download:
|
| Fig. 11. Kinetics studies of n-hexylamine-initiated Bn-Glu NCA ROP at room temperature with Bn-Glu NCA/initiator ratio of 100 : 1. (a) The initial Bn-Glu NCA concentration was 50.0 mg/mL (0.19 mmol/mL). The N2 flow rates were 0 mL/min (green line), 100 mg/mL (black line), and 250.0 mg/mL (red line). (b). NCA ROP terminated at selected monomer conversion with a flow rate of 100.0 mg/mL. Reproduced with permission [67]. Copyright 2013, American Chemical Society. | |
Stukenkemper et al. reported another interesting unique strategy to synthesize polypeptides [69]. They published the first example of ultraviolet (UV)-initiated propagation of NCAs. The preinitiator photoamine would dissociate in situ under the UV exposure so that the selective initiation process was accessible. The proposed photoinitiation mechanism was involved with the attachment of the active initiators to the polypeptide chains according to the NAM as in traditional NCA polymerizations with the same initiators (Fig. 12). Although it was difficult to ascertain the ratio of monomer to the initiator in the photoinitiation process under the conditions applied, the molecular weights of products could be controlled through regulating the light introduced to the system either by varying the irradiation time or the distance between lamp and reactor. Moreover, the polymers were structurally well-defined. The work might particularly offer fascinating opportunities for surface-initiated processes with possibilities for spatial polymerization control.

|
Download:
|
| Fig. 12. Photoinitiation mechanism. Reproduced with permission [69]. Copyright 2017, Royal Society of Chemistry | |
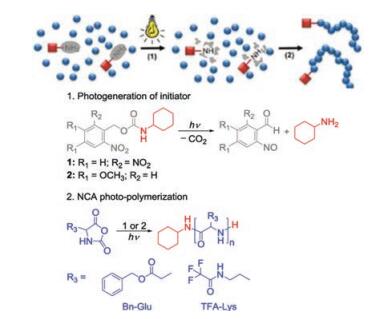
Without addition of any initiator or catalyst, Dong's group combined photocaged chemistry and the inimer (initiator + monomer) ROP to prepare linear polypeptides with high molecular weights (109 kDa) or hyperbranched polypeptides (HBPPs) with tunable molecular weights (7.6–90.8 kDa) and degree of branching (DB) (0.09–0.60) [70]. To further optimize this reaction in terms of shortening the UV-irradiation period and restraining the side reactions through deactivation of photo-byproduct, a new photocaged α-amino acid monomer N(ε)-(1-(2-nitrophenyl) ethoxycarbonyl)-L-lysine NCA (NPEOC-Lys NCA) was designed [71]. In addition, the authors innovatively combined the UV light and N2 flow during propagation. Through tuning the UV irradiation intensity and/or the N2 flow rate, lysine-based HBPPs with tunable DB (0.21–0.54) and higher actual molecular weights (13.7–62.8 kDa) could be efficiently produced.

|
Download:
|
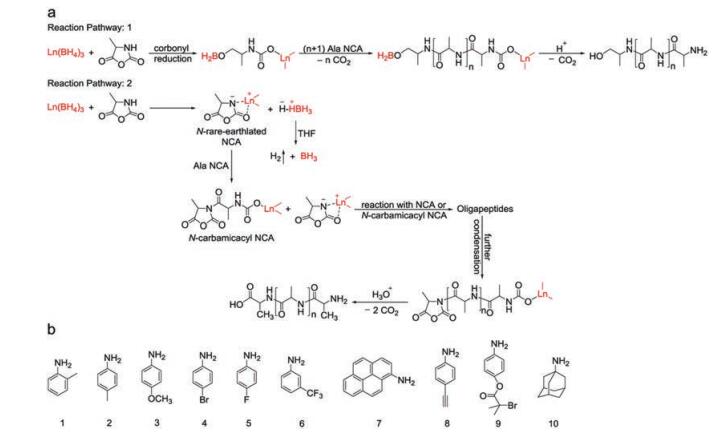
| Fig. 13. Metallic catalysts and corresponding mechanism. (a) Mechanism for ROP of Ala NCA catalyzed by Ln(BH4)3(THF)3. Reproduced with permission [73]. Copyright 2012, John Wiley & Sons, Inc. (b) Chemical structures of investigated aniline analogs. Reproduced with permission [76]. Copyright 2018, MDPI. | |
Besides the optimization of initiators and experimental conditions, catalysts also play significant roles in the controlled ROP of NCAs. In contrast to several reports in terms of initiating systems, fewer work of catalysts have been published, as presented in Table 2. Many kinds of metallic complexes are able to act as effective catalysts for the ROP of NCAs, including alkali metal compounds, rare earth metal compounds, transition mental compounds, and so forth. Through participating in reaction pathways, catalysts are able to promote the controllable synthesis of polypeptides and shorten the propagation time in high yield.
|
|
Table 2 Catalysts for controlled polymerization of α-amino acid NCAs. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In 2011, a series of rare earth complexes were first introduced by Shen and coworkers as potent catalysts for the ROP of Bn-Glu NCA and L-alanine NCA (Ala NCA), including rare earth isoprop-oxide (RE(OiPr)3), rare earth tris(2, 6-di-tert-butyl-4-methylphenolate) (RE(OAr)3), rare earth tris (borohydride) (RE(BH4)3(THF)3), rare earth tris[bis(trimethylsilyl)amide] (RE(NTMS)3), and rare earth trifluoromethanesulfonate [72]. Their structural formulas are presented in Table 2 in order. The first four showed high catalytic activities toward the NCA polymerizations, and the produced polypeptides were in high yields (> 90%) with moderate PDIs ranging from 1.2 to 1.6. Polymerization features and chain structures of these five catalytic systems were studied in comparison.
On the basis of previous work, the authors further synthesized homo, random, and block copolymers in high yield with expected molecular weights and relatively low PDIs (1.1–1.6) by Ln(BH4)3(THF)3 (Ln = Sc, Y, La, and Dy), confirming the livingness of the polymerization. The molecular weights of homopolypeptides could be controlled by the molar ratios of monomer to the catalyst. According to the proposed mechanism, NAM and AMM were concurrent during propagation, which led to the coexistence of two active centers. The nucleophilic attack by the rare earth catalysts on the 5-CO and deprotonation of 3-NH simultaneously took place in the initiation process [73]. Propagation then proceeded on active centers via both regular monomer insertion and polycondensation, as displayed in Fig. 13a.
It is reported that sodium hydroxide (NaOH), sodium methoxide (NaOCH3), sodium borohydride (NaBH4), and triethylamine (N(C2H5)3) mediated polymerization of Bn-Glu NCA as early as 1956. They generated PBLG with molecular weights over 500, 000 (DP > 2000) [74]. NaBH4, as a commercially-available compound, however, has not been further investigated in NCA polymerization thereafter. In 2014, Ling's group reported alkali metal borohydrides (MBH4, M = Li, Na, and K) as catalysts for the ROP of NCAs producing α-hydroxy-ω-aminotelechelic polypeptides, which were prepared in quantitative yields with relatively narrow molecular weight distributions (PDIs = 1.1–1.5) depending on reaction temperature [75]. Compared to the alkyl and alkoxyl end groups, the hydroxyl end group provided an available site for post-modification functionalization.
Transition metal complexes also provide platforms for the convenient synthesis of highly desirable polypeptides. Lately, an easily accessible catalyst system composed of zinc acetate (Zn(OAc)2) and aniline derivates was explored by Nie et al. to promote the fast polymerization of Bn-Glu NCA [76]. It was demonstrated that such a combination generated PBLG with controllable molecular weight and narrow PDI within relatively short polymerization time. Moreover, thanks to the good compatibility of Zn(OAc)2 with a broad range of aniline analogs, their combinations provided a promising platform for easily obtaining end-functionalized PBLG and block copolymers to satisfy the requirements of numerous biological applications further. The chemical structures of aniline analogs adopted in this study are presented in Fig. 13b.
4.2. OrganocatalystsCompared with metallic catalysts, metal-free catalysts are preferable as it can produce polymers without any metallic residues, providing satisfactory materials for demanding and sensitive domains, such as biomedical applications, microelectronic devices, and food packaging. Based on alcohol/thiourea hydrogen-bonding function, Zhao et al. devised and reported a unique catalyst, namely N, N'-bis[3, 5-bis(trifluoromethyl)phenyl] thiourea (TU-S), that offered an effective platform for fast controlled ROP of α-amino acids NCAs using amino alcohol as initiators under mild conditions in 2015 [77]. The structures of amino alcohol initiators are listed in Fig. 14a. The TU-S simultaneously activated the NCA monomer, reversibly neutralized the growing chain ends and disabled the tertiary amine, which suppressed the unwanted side products generated by AMM (Fig. 14b), and thus realized a controlled polymerization. For instance, by employing N, N-dimethyl ethanolamine as an initiator in the presence of TU-S, a spectrum of well-defined linear polypeptides with differently designed molecular weights (3.01 × 104– 18.10 × 104) and low PDI values (1.02–1.05) were easily achieved.

|
Download:
|
| Fig. 14. Organocatalytic systems and corresponding mechanisms. (a) Initiators used in alcohol/thiourea hydrogen-bonding organocatalytic systems. (b) Reaction mechanism of living ROP catalyzed by TU-S. Reproduced with permission [77]. Copyright 2015, Royal Society of Chemistry. (c) Proposed mechanism for ROP of BnGlu NCA in the (S)-1, 10-binaphthyl-based phosphoric acid catalytic system. Reproduced with permission [79]. Copyright 2019, Elsevier Ltd. | |
{kind=link}
Moreover, this strategy was introduced successfully towards the preparation of well-defined (co)polypeptides bearing pendant alkyne groups in 2016 by Hadjichristidis's group [78]. For the first time, it was demonstrated that amino alcoholsin the presence of TUS could function as potent initiators for the fast living ROP of propargyl-Glu NCA under mild conditions. This method generated a series of well-defined di-/multi-armed alkyne functionalized homo-/co-polypeptides, such as AB diblock (B: non-functionalized), ABA triblock, and star-AB diblock. Furthermore, linear and star random co-polypeptides were prepared as precursors of a number of complex macromolecular architectures by click chemistry.
More recently, accompanied by changing the highly tunable substituent groups at 2, 20-position of the binaphthyl skeleton, (R)- and (S)-binaphthyl phosphoric acids were respectively utilized to catalyze the ROP of NCAs by Liang et al. [79]. They aimed to investigate the possible effect of the optical configurations of catalytic systems on the controllability of polymerization. Consequently, (S)-1, 10-binaphthyl-2, 20-dimethoxyl phosphate was confirmed to efficiently promote the ROP of Bn-Glu NCA initiated by various primary amines, fabricating perfect linear PBLG with controlled molecular weight and narrow dispersity in relatively short polymerization time. In the meanwhile, a plausible mechanism involving primary amine-phosphoric acids mode was proposed in the (S)-1, 10-binaphthyl-based phosphoric acid catalytic system, as shown in Fig. 14c.
Fluorinated alcohol-based catalytic system for the ROP of α-amino acid NCAs, namely 1, 3-bis(2-hydroxy hexafluoroisopropyl) benzene, characterized by cocatalyst free, metal free, high rate, and high selectivity, was reported by Zhao et al. [80]. It is the multiple dynamic hydrogen bonds formed between the fluorinated alcohols and the initiators, monomers, and propagating polymer chains during polymerization, which activate the NCAs and simultaneously protect the overactive polymer chain ends. In this way, the high activity and selectivity of propagation are accessible.
In 2017, Zhang et al. put forward a concept of frustrated Lewis pairs (FLPs), which was comprised of bulky borane Lewis acid and amine group. The nucleophilic Lewis base in FLPs still functioned as an initiator during polymerization. Thanks to the continuous formation of FLP intermediate between bulky Lewis acid and ω-amine end of the propagating chain, the side reactions were efficiently restrained so that the polypeptides with well-defined chemical structures could be synthesized under mild condition [81].
5. Conclusion and perspectivesThanks to the diversity of chemical structure, different scales of devices, and remarkable biocompatibility, as well as biodegrad-ability of polypeptides, controlled synthesis of polypeptides by simple chemical techniques is a critical subject for their biomedical applications. Beyond all doubt, the ROP of α-amino acid NCAs is an essential strategy for the preparation of polypeptides. However, the uncontrollable aspects of ROP hinder accurate polypeptide synthesis and their further applications in biomedical engineering (drug delivery, gene delivery, and tissue engineering) [33] and analysis (biosensors and medical diagnosis) [82]. Firstly, α-amino acid NCA is vulnerable to moisture, so it is necessary to polymerize in glovebox to maintain an anhydrous condition. Secondly, because of the concurrency of AMM and NAM during polymerization, the obtained products are plagued with reduced control over molecular weights, wide PDIs, and imprecise sequences. Furthermore, the possible side reactions, including undesired chain transfer and indispensable chain termination reactions, would also restrict the synthesis of well-defined polypeptide architectures. Thirdly, slow polymerization kinetics induces long reaction time. To sum up, it is necessary and vital to explore fast and living ROP strategies to obtain polypeptide architectures with variable chemical compositions and topologies, high molecular weights, as well as narrow PDIs.
Much attention of scientists has been attracted. Thus, recent several decades have witnessed the steady progression in terms of the precise synthesis of polypeptides with different topologies and compositions. In this review, we offered a general overview in respect of the controlled polymerization strategies, including the development of effective initiating systems, the optimization of reaction conditions, and the exploration of promising catalysts. Initiating systems play vital roles during ROP reactions, which can be divided into four categories according to their composition: metal-based complexes, primary amine hydrochloride salts, silane derivates, and other initiating compounds. Reaction conditions, regarding high pressure, low temperature, N2 atmosphere, etc., would also facilitate the controlled ROP. Besides, metallic catalysts and organocatalysts provide promising platforms for precise polypeptide synthesis. These improvements largely optimize the reaction process, such as shortening polymerization time, increasing monomer conversion rate, and restraining the possible side reactions. So they expand the scope of available artificial polypeptides, which possess various physical and chemical properties. Then the polypeptide architectures would self-assemble and form into nanoparticles, such as vesicles, micelles, nanogels, or macroscopic hydrogels, to function in various biomedical applications [3, 83].
The various synthetic methods provide chances to tailor products in order to meet the specific requirements of specific biomedical or pharmaceutical needs. Despite these accomplishments, there is still much effort need to pay in the following two aspects. On the one hand, in terms of the strategies published yet, it is essential to explore their scopes of applications, namely which kinds of α-amino acid NCA can form polymers close to the ideal ones. The researchers should investigate as detailed as possible to facilitate their wide applications in the synthesis of functionalized polypeptides with well-defined structures. Furthermore, for some methods whose mechanisms are ambiguous, further study should be committed to revealing their concrete reaction pathways, such as solubility and chemical structures of the growing peptide chains, termination reactions, and so forth. Moreover, researches should be devoted to a detailed kinetic analysis. Only in this way can we find out the influence of experimental parameters on target product properties in order to achieve a living ROP ultimately. On the other hand, designing unique synthetic approaches of polypeptides, which are similar to the natural counterparts in terms of complexity and accuracy in a fast and controlled manner, are still a significant project. Enhancing the polymerization kinetics and expanding the scope of polypeptides are meaningful. In addition, it is vital to develop advanced initiating systems enabling the formation of functionalizing polypeptides and polypeptide hybrid copolymers.
AcknowledgmentsThe authors are grateful for the financial support from the National Natural Science Foundation of China (Nos. 51973216, 51873207, 51803006, 51833010, 51673190 and 51520105004), the Science and Technology Development Program of Jilin Province (No. 20190201068JC), the National Key Research and Development Program of China (No. 2016YFC1100701), the Youth Talents Promotion Project of Jilin Province (No. 181909), and the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2019005).
| [1] |
Z. Song, Z. Han, S. Lv, et al., Chem. Soc. Rev. 46 (2017) 6570-6599. DOI:10.1039/C7CS00460E |
| [2] |
Z. Song, Z. Tan, J. Cheng, Macromolecules 52 (2019) 8521-8539. DOI:10.1021/acs.macromol.9b01450 |
| [3] |
C. Deng, J. Wu, R. Cheng, et al., Prog. Polym. Sci. 39 (2014) 330-364. DOI:10.1016/j.progpolymsci.2013.10.008 |
| [4] |
H. Tian, Z. Tang, X. Zhuang, X. Chen, X. Jing, Prog. Polym. Sci. 37 (2012) 237-280. DOI:10.1016/j.progpolymsci.2011.06.004 |
| [5] |
T.J. Deming, Prog. Polym. Sci. 32 (2007) 858-875. DOI:10.1016/j.progpolymsci.2007.05.010 |
| [6] |
H. Lu, J. Wang, Z. Song, et al., Chem. Commun. 50 (2014) 139-155. DOI:10.1039/C3CC46317F |
| [7] |
C. He, X. Zhuang, Z. Tang, H. Tian, X. Chen, Adv. Healthc. Mater. 1 (2012) 48-78. DOI:10.1002/adhm.201100008 |
| [8] |
R. Duncan, Nat. Rev. Drug Discov. 2 (2003) 347-360. DOI:10.1038/nrd1088 |
| [9] |
T.J. Deming, Adv. Mater. 9 (1997) 299-311. DOI:10.1002/adma.19970090404 |
| [10] |
H.A. Klok, Macromolecules 42 (2009) 7990-8000. DOI:10.1021/ma901561t |
| [11] |
B. Bacsa, K. Horváti, S. Bõsze, F. Andreae, C.O. Kappe, J. Org. Chem. 73 (2008) 7532-7542. DOI:10.1021/jo8013897 |
| [12] |
N. Hadjichristidis, H. Iatrou, M. Pitsikalis, G. Sakellariou, Chem. Rev. 109 (2009) 5528-5578. DOI:10.1021/cr900049t |
| [13] |
H.R. Kricheldorf, Angew. Chem. Int. Ed. 45 (2006) 5752-5784. DOI:10.1002/anie.200600693 |
| [14] |
H. Leuchs, Ber. Dtsch. Chem. Ges. 39 (1906) 857-861. DOI:10.1002/cber.190603901133 |
| [15] |
H. Leuchs, W. Manasse, Ber. Dtsch. Chem. Ges. 40 (1907) 3235-3249. DOI:10.1002/cber.19070400387 |
| [16] |
H. Leuchs, W. Geiger, Ber. Dtsch. Chem. Ges. 41 (1908) 1721-1726. DOI:10.1002/cber.19080410232 |
| [17] |
T. Curtius, W. Sieber, Ber. Dtsch. Chem. Ges. 54 (1921) 1430-1437. DOI:10.1002/cber.19210540707 |
| [18] |
T. Curtius, W. Sieber, Ber. Dtsch. Chem. Ges. 55 (1922) 1543-1558. DOI:10.1002/cber.19220550608 |
| [19] |
T. Curtius, J. Prakt. Chem. 125 (1930) 211-218. DOI:10.1002/prac.19301250110 |
| [20] |
F. Wessely, Hoppe-Seyler's Z. Physiol. Chem. 146 (1925) 72-90. DOI:10.1515/bchm2.1925.146.1-3.72 |
| [21] |
F. Wessely, J. Mayer, Monatsh. Chem. Verw. Teile Anderer Wiss. 50 (1928) 439-448. DOI:10.1007/BF01552465 |
| [22] |
F. Wessely, K. Riedl, H. Tuppy, Monatsh. Chem. 81 (1950) 861-872. DOI:10.1007/BF00899328 |
| [23] |
F. Wessely, W. Swoboda, Monatsh. Chem. 82 (1951) 621-627. DOI:10.1007/BF00902814 |
| [24] |
K. Schlogl, F. Wessely, G. Korger, Monatsh. Chem. 83 (1952) 845-864. DOI:10.1007/BF00899623 |
| [25] |
G.J.M. Habraken, M. Peeters, C.H.J.T. Dietz, C.E. Koning, A. Heise, Polym. Chem. 1 (2010) 514-524. DOI:10.1039/b9py00337a |
| [26] |
T. Curtius, K. Hochschwender, H. Meier, et al., J. Prakt. Chem. 125 (1930) 211-302. DOI:10.1002/prac.19301250110 |
| [27] |
H.R. Kricheldorf, C. von Lossow, G. Schwarz, Macromolecules 38 (2005) 5513-5518. DOI:10.1021/ma0401368 |
| [28] |
M. Barz, R. Luxenhofer, R. Zentel, M.J. Vicent, Polym. Chem. 2 (2011) 1900-1918. DOI:10.1039/c0py00406e |
| [29] |
H.R. Kricheldorf, C. Von Lossow, G. Schwarz, Macromol. Chem. Phys. 206 (2005) 282-290. DOI:10.1002/macp.200400417 |
| [30] |
I. Dimitrov, H. Kukula, H. Colfen, H. Schlaad, Macromol. Symp. 215 (2004) 383-393. DOI:10.1002/masy.200451129 |
| [31] |
T.J. Deming, Nature 390 (1997) 386-389. DOI:10.1038/37084 |
| [32] |
C.M. Gonzalez-Henriquez, M.A. Sarabia-Vallejos, J. Rodriguez-Hernandez, Polymers 9 (2017) 551-612. DOI:10.3390/polym9110551 |
| [33] |
P. Zheng, Y. Liu, J. Chen, et al., Chin. Chem. Lett. 31 (2020) 1178-1182. DOI:10.1016/j.cclet.2019.12.001 |
| [34] |
T.J. Deming, Macromolecules 32 (1999) 4500-4502. DOI:10.1021/ma9902899 |
| [35] |
T.J. Deming, S.A. Curtin, J. Am. Chem. Soc. 122 (2000) 5710-5717. DOI:10.1021/ja994281q |
| [36] |
S.A. Curtin, T.J. Deming, J. Am. Chem. Soc. 121 (1999) 7427-7428. DOI:10.1021/ja990905g |
| [37] |
Y.L. Peng, S.L. Lai, C.C. Lin, Macromolecules 41 (2008) 3455-3459. DOI:10.1021/ma7025836 |
| [38] |
A.A. Goodwin, X.H. Bu, T.J. Deming, J. Organomet. Chem. 589 (1999) 111-114. DOI:10.1016/S0022-328X(99)00299-5 |
| [39] |
S.W. Seidel, T.J. Deming, Macromolecules 36 (2003) 969-972. DOI:10.1021/ma025844c |
| [40] |
A. Bhaw-Luximon, D. Jhurry, J. Belleney, V. Goury, Macromolecules 36 (2003) 977-982. DOI:10.1021/ma0214310 |
| [41] |
I. Dimitrov, H. Schlaad, Chem. Commun. 23 (2003) 2944-2945. |
| [42] |
I. Conejos-Sánchez, A. Duro-Castano, et al., Polym. Chem. 4 (2013) 3182-3186. DOI:10.1039/c3py00347g |
| [43] |
C.D. Vacogne, H. Schlaad, Chem. Commun. 51 (2015) 15645-15648. DOI:10.1039/C5CC06905J |
| [44] |
H. Lu, J. Cheng, J. Am. Chem. Soc. 129 (2007) 14114-14115. DOI:10.1021/ja074961q |
| [45] |
Y.M. Wu, D.F. Zhang, P.C. Ma, et al., Nat. Commun. 9 (2018) 5297-5306. DOI:10.1038/s41467-018-07711-y |
| [46] |
H. Lu, J. Cheng, J. Am. Chem. Soc. 130 (2008) 12562-12563. DOI:10.1021/ja803304x |
| [47] |
H. Lu, J. Wang, Y. Lin, J. Cheng, J. Am. Chem. Soc. 131 (2009) 13582-13583. DOI:10.1021/ja903425x |
| [48] |
J. Yuan, Y. Sun, J. Wang, H. Lu, Biomacromolecules 17 (2016) 891-896. DOI:10.1021/acs.biomac.5b01588 |
| [49] |
Y. Hou, J. Yuan, Y. Zhou, J. Yu, H. Lu, J. Am. Chem. Soc. 138 (2016) 10995-11000. DOI:10.1021/jacs.6b05413 |
| [50] |
Y. Hou, Y. Zhou, H. Wang, et al., J. Am. Chem. Soc. 140 (2018) 1170-1178. DOI:10.1021/jacs.7b13017 |
| [51] |
J.S. Yuan, Y. Zhang, Z.Z. Li, Y.Y. Wang, H. Lu, ACS Macro Lett. 7 (2018) 892-897. DOI:10.1021/acsmacrolett.8b00465 |
| [52] |
W. Zhao, Y. Gnanou, N. Hadjichristidis, Biomacromolecules 16 (2015) 1352-1357. DOI:10.1021/acs.biomac.5b00134 |
| [53] |
W. Zhao, Y. Gnanou, N. Hadjichristidis, Chem. Commun. 51 (2015) 3663-3666. DOI:10.1039/C4CC09055A |
| [54] |
S. Gradisar, E. Zagar, D. Pahovnik, ACS Macro Lett. 6 (2017) 637-640. DOI:10.1021/acsmacrolett.7b00379 |
| [55] |
X. Zhang, M. Oddon, O. Giani, S. Monge, J.J. Robin, Macromolecules 43 (2010) 2654-2656. DOI:10.1021/ma9025916 |
| [56] |
T. Aliferis, H. Iatrou, N. Hadjichristidis, Biomacromolecules 5 (2004) 1653-1656. DOI:10.1021/bm0497217 |
| [57] |
T. Aliferis, H. Iatrou, N. Hadjichristidis, J. Polym. Sci. Part A:Polym. Chem. 43 (2005) 4670-4673. DOI:10.1002/pola.20926 |
| [58] |
A. Karatzas, H. Iatrou, N. Hadjichristidis, et al., Biomacromolecules 9 (2008) 2072-2080. DOI:10.1021/bm800316w |
| [59] |
D.L. Pickel, N. Politakos, A. Avgeropoulos, J.M. Messman, Macromolecules 42 (2009) 7781-7788. DOI:10.1021/ma901340y |
| [60] |
W. Vayaboury, O. Giani, H. Cottet, A. Deratani, F. Schue, Macromol. Rapid Commun. 25 (2004) 1221-1224. DOI:10.1002/marc.200400111 |
| [61] |
W. Vayaboury, O. Giani, H. Cottet, S. Bonaric, F.J.M.C. Schué, Physics 209 (2008) 1628-1637. |
| [62] |
G.J.M. Habraken, M. Peeters, C.H.J.T. Dietz, C.E. Koning, A. Heise, Polym. Chem. 1 (2010) 514-524. DOI:10.1039/b9py00337a |
| [63] |
G.J.M. Habraken, K.H.R.M. Wilsens, C.E. Koning, A. Heise, Polym.Chem. 2 (2011) 1322-1330. DOI:10.1039/c1py00079a |
| [64] |
H. Cao, J.R. Yao, Z.Z. Shao, Polym. Int. 61 (2012) 774-779. DOI:10.1002/pi.4138 |
| [65] |
X.C. Li, C.S. Hu, H.J. Li, et al., Polymer 151 (2018) 1-5. DOI:10.1016/j.polymer.2018.07.053 |
| [66] |
D. Thunig, J. Semen, H.G. Elias, Makromol. Chem. 178 (1977) 603-607. DOI:10.1002/macp.1977.021780229 |
| [67] |
J. Zou, J.W. Fan, X. He, et al., Macromolecules 46 (2013) 4223-4226. DOI:10.1021/ma4007939 |
| [68] |
J. Muzart, Tetrahedron 65 (2009) 8313-8323. DOI:10.1016/j.tet.2009.06.091 |
| [69] |
T. Stukenkemper, J. Jansen, C. Lavilla, et al., Polym. Chem. 8 (2017) 828-832. DOI:10.1039/C6PY02018F |
| [70] |
P. Li, C.M. Dong, ACS Macro Lett. 6 (2017) 292-297. DOI:10.1021/acsmacrolett.7b00167 |
| [71] |
P. Li, Y. Song, C.M. Dong, Polym. Chem. 9 (2018) 3974-3986. DOI:10.1039/C8PY00641E |
| [72] |
H. Peng, J. Ling, Z. Shen, J. Polym. Sci. Part A:Polym. Chem. 50 (2012) 1076-1085. DOI:10.1002/pola.25848 |
| [73] |
H. Peng, J. Ling, Y. Zhu, L. You, Z. Shen, J. Polym. Sci. Part A:Polym. Chem. 50 (2012) 3016-3029. DOI:10.1002/pola.26077 |
| [74] |
E.R. Blout, R.H. Karlson, J. Am. Chem. Soc. 78 (1956) 941-946. DOI:10.1021/ja01586a020 |
| [75] |
H. Peng, W.L. Chen, J. Kong, Z.Q. Shen, J. Ling, Chin. J. Polym. Sci. 32 (2014) 743-750. DOI:10.1007/s10118-014-1445-6 |
| [76] |
Y.Z. Nie, X.M. Zhi, H.F. Du, J. Yang, Molecules 23 (2018) 760-772. DOI:10.3390/molecules23040760 |
| [77] |
W. Zhao, Y. Gnanou, N. Hadjichristidis, Polym. Chem. 6 (2015) 6193-6201. DOI:10.1039/C5PY00874C |
| [78] |
W. Zhao, Y. Gnanou, N. Hadjichristidis, Polym. Chem. 7 (2016) 3487-3491. DOI:10.1039/C6PY00365F |
| [79] |
J. Liang, X. Zhi, Q. Zhou, J. Yang, Polymer 165 (2019) 83-90. DOI:10.1016/j.polymer.2019.01.027 |
| [80] |
W. Zhao, Y. Lv, J. Li, et al., Nat. Commun. 10 (2019) 3590-3600. DOI:10.1038/s41467-019-11524-y |
| [81] |
H. Zhang, Y. Nie, X. Zhi, H. Du, J. Yang, Chem. Commun. 53 (2017) 5155-5158. DOI:10.1039/C7CC01440F |
| [82] |
J.N. Cha, G.D. Stucky, D.E. Morse, T.J. Deming, Nature 403 (2000) 289-292. DOI:10.1038/35002038 |
| [83] |
Z. Zheng, L. Zhang, Y. Ling, H. Tang, Eur. Polym. J. 115 (2019) 244-250. DOI:10.1016/j.eurpolymj.2019.03.034 |