 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Fine Chemicals, Institute of Artificial Photosynthesis, Dalian University of Technology, Dalian 116024, China
Due to the long-lived property, the triplet excitons of photoresponsive materials are extremely useful in the applications of photodynamic therapy [1-3], photocatalysis [4-6], triplet-triplet annihilation photon up-conversion [7-9], light-emitting diodes [10, 11], and photovoltaics [12, 13]. Especially in the photovoltaic field, the lifetime of excitons directly determines their diffusion lengths, and thereby affecting the power conversion efficiency (PCE) of the device. The singlet excitons with the short lifetime are easily quenched and thereby transporting less than 10 nm, while the triplet ones can overcome these disadvantages and increase the diffusion length up to 10 μm [14]. In the cases of perovskite and silicon solar cells, due to the long lifetimes of the excitons, it is possible to increase the thickness of the active layer more than 1 μm to gain enough solar photons and have achieved the PCEs over 25% [13, 15]. Generally, for the organic dyes, the intersystem crossing (ISC) process is spin-forbidden, and usually quantum yield is quite low. Chemists have developed several ways to enhance ISC, such as introducing the heavy metal atom in the molecules [16-18], linking the triplet photosensitizer [19-21] and constructing the electron donor-acceptor (D-A) structures [22-24]. Besides these, extending the non-planar π-conjugation is found to facilitate ISC via strengthening the spin-orbit coupling [25].
Perylene diimides (PDI) dyes are known as photothermally stable compounds. They have strong absorption in the visible and controllable electron energy levels. Therefore, they are one of the most important electron acceptor materials in the non-fullerene organic solar cells (OSCs) [26-30]. However, for the PDI monomer, the large phase separation strongly decrease the exciton separation efficiency [31]. To inhibit the intermolecular aggregation, there are lots of twisted PDI derivatives created for high PCEs [32-37]. Among them, the bay-position derived PDI dimers, constructed by a donoracceptor structure, are designed to control the phase separation domain and greatly improve the efficiencies of the OSCs [34, 35, 38-40]. An interesting result is that the best PCEs of the PDI dimers could be promoted again afterforming the ring-fused structure [35, 38, 39]. For example, in the previous publications [38, 39], two thiophenecoupled PDI dyads (as shown in Fig. 1, ring-unfused2PDI-Thand ringfused fused-2PDI-Th) as electron acceptor materials in OSC devices were tested under the same conditions. The PCEs of OSC based on 2PDI-Th and fused-2PDI-Th are 3.65% and 6.72% [38], (or 2.19% and 3.44% [39]) respectively. The improved device performance was attributed to the appropriate phase separation domain, high and balanced carrier mobility, raised lowest unoccupied molecular orbital(LUMO)levels, and decreased geminate recombination.These explanations are undoubtedly reasonable from their individual perspectives of OSC devices. However, it is necessary to consider the differences in the properties of excitation states (especially long life triplet excitons, as mentioned previously) for the two electron acceptors of the unfused and fused PDI dimers.
Looking at the unfused and fused molecular structures with two identical PDI units, it is logical to assume that there exist crucial differences in excited state photophysical processes and lifetimes of the excitons. They control whether the excitons can reach at the interface for charge separating to achieve high short circuit currents (JSC), because an obvious fact is that the devices based on ring-fused compounds gave higher JSC than those of the ringunfused ones in the bulk-heterojunction OSCs [38, 39]. Therefore, we desire to investigate their intrinsic properties of the singlet transition processes and exciton lifetimes of this typed PDI dimers, which are never be considered.
Herein, 2PDI-Th and fused-2PDI-Th have been investigated by the ultrafast transient absorption (TA) spectroscopy. As a result, the two compounds exhibit absolutely different singlet excited state relaxation dynamics and triplet excited lifetimes of the excitons. The singlet excited excitons of 2PDI-Th firstly format the PDI excimers within 1.5 ps, and then the triplet state is generated from these excimers via intersystem crossing (ISC) within 0.5–1.1 ns, while the singlet excited excitons of fused-2PDI-Th directly format the triplet excited state with a generation time of 3.3 ns. And the triplet exciton lifetime of fused-2PDI-Th is about 67 μs, which is over 3 times longer than that (22 μs) of 2PDI-Th. All these results indicate that such excitons of fused-2PDI-Th, with not excimer formation but long lifetime, could diffuse in a long distance, which are beneficial for achieving high PCEs in OSCs.
The two compounds were synthesized according to the previous publications [38, 39]. To understand the photophysical processes of the two compounds, the parent PDI monomer (Fig. 1) was also measured as a reference. All the compounds were fully characterized with NMR and MS spectra (Supporting information). The ground-state geometries and the corresponding energy levels are optimized by the density functional theory (DFT) calculations using the B3LYP functional and a 6-31G(d, p) basis set. The excitedstate geometries and the corresponding energy levels are simulated by TD-DFT with the same basis set as that of DFT.

|
Download:
|
| Fig. 1. Molecular structures of PDI, 2PDI-Th and fused-2PDI-Th. | |
{kind=link}
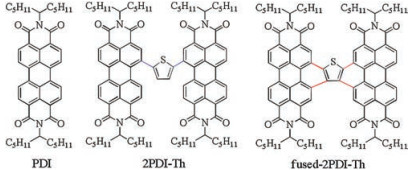
As shown in Fig. 2a, the UV–vis absorption spectra was measured in dilute dichloromethane with a concentration of 1 ×10-5 mol/L. Compared with PDI, 2PDI-Th exhibits a broad and red-shifted absorption band, while fused-2PDI-Th shows a narrow and blue-shifted spectra with an increased absorbance in the higher energy region due to the rigid thiophene fused π-conjugation. All three compounds show strong visible absorption with the maximum molar extinction coefficients of 72, 000, 61, 000 and 68, 000 L mol-1 cm-1 for PDI, 2PDI-Th and fused-2PDI-Th, respectively. As shown in Fig. 2b, an analogous trend was found in the fluorescence spectra of fused-2PDI-Th with the emission maximum of 540 nm, slight red-shift compared with that of PDI. From the emission spectra, we see clearly that the fluorescence intensities are different, which indicates very different fluorescence yields in three compounds. For 2PDI-Th, it emits relatively strong fluorescence in the solution with an absolute fluorescence quantum yield (ΦF) of 0.17. The fluorescence spectra of 2PDI-Th becomes very weak (with a low ΦF of 0.02), broader and featureless with strong red-shifted emission peak at 674 nm, which is the conspicuous feature of excimers [41-44]. In addition, the excimer formation is well-known as a trapping exciton process and has a serious impact on exciton transporting [31, 45-49]. Although the reported structures that can easily form the excimer state excitons are the face-to-face geometry via N-positionconnected PDI dimmers [42, 42, 43, 44, 50], we believe that 2PDI-Th derived from the bay-position connection may also get into this state.

|
Download:
|
| Fig. 2. Steady-state absorption (a) and emission (b) spectra (inset with small scale of y-axes) in dichloromethane of PDI, 2PDI-Th, fused-2PDI-Th; Optimized ground- (c, e) and excited-state (d, f) geometries for 2PDI-Th and fused-2PDI-Th, respectively. | |
{kind=link}
We simulated the molecular geometry using DFT and TD-DFT calculations. As shown in Fig. 2c, the optimized ground state (S0) configuration of 2PDI-Th is a twisted structure with a dihedral angle of 72° between the two separated PDI units. In their optimized first excited singlet state (S1) (Fig. 2d), the dihedral angle is decreased to 42°. Although two PDI monomers are not bound in the face-to-face geometry, especially in excited state, they are similar to an H-configuration. Thus, this molecular configuration is beneficial to form the excimer state. Furthermore, it is commonly known that molecular rotations often dissipate the excited state energy through the exothermic way and therefore quenching the fluorescence of dyes [51]. Indeed, from S0 to S1 of 2PDI-Th, the dihedral angle has changed of 30°, which can explain the extremely low fluorescence quantum yield. We conclude that the steadystate emission spectra is attributed to the excimer excitons. For fused-2PDI-Th, in spite of the enhanced planarity and rigidity of the molecule by ring fusion, there is still a twist angle of 29° between the two separated PDI units in the optimized S0 state (Fig. 2e). In the optimized S1 state (Fig. 2f), the molecular configuration is nearly the same as that of S0 with a twist angle of 28°. The entire molecule of fused-2PDI-Th exhibits an X-shaped configuration, which is different from that of 2PDI-Th. Therefore, the DFT and TD-DFT calculations reveal that the different molecular configurations determine their unique steady-state spectra properties.
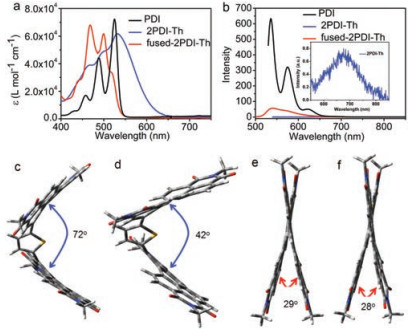
The fluorescence maps of PDI, 2PDI-Th and fused-2PDI-Th in dichloromethane were measured by time-resolved single photon counting (TCSPC) technique at λex = 400 nm (Fig. 3). For PDI (Fig. 3a), the fluorescence at 520-650 nm, corresponds well with the steady-state fluorescence spectrum and exhibits a monoexponential decay with a lifetime of 5.5 ns (Table S1 in Supporting information), in agreement with the publication [52]. However, 2PDI-Th exhibits different emission properties (Fig. 3b). The fluorescence is red shifted (550-780 nm), moreover it decays biexpontially with major component of 840 ps and minor contribution of 100 ps (Table S2 in Supporting information). We assign this red shifted emission to fluorescence of excimer. It should be mentioned that the weak fluorescence at 500-550 nm is due to remaining impurities of fused-2PDI-Th, since their decay times are similar, comparing 3.5 ns with 3.3 ns of fused-2PDI-Th (Tables S2 and S3 in Supporting information). We have also observed that 2PDI-Th is photochemically highly sensitive, i.e., easily can be transferred into fused-2PDI-Th. For fused-2PDI-Th, the TCSPC map spectrum corresponds well with the steady-state fluorescence spectrum (compare Figs. 2b and 3 c). And from the deconvolution results, the fluorescence exhibits monoexponential decay with the lifetime of 3.3 ns (Table S3).

|
Download:
|
| Fig. 3. TCSPC maps of (a) PDI, (b) 2PDI-Th, and (c) fused-2PDI-Th in 1 cm cuvette, excitation at 400 nm. | |
{kind=link}
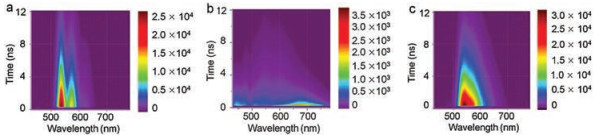
For further detailed study of photochemical and photophysical processes, the femtosecond transient absorption (fsTA) technique were applied with excitation wavelength of 500 nm (Fig. 4). The fsTA spectra of PDI are presented in Fig. 4a. Excited-state absorption (ESA) band was observed at 700 nm, which is assigned to singlet-singlet absorption with decay lifetime of 5.3 ns (Fig. 4b and Table S4 in Supporting information), in agreement with the time-resolved fluorescence data and that from the publication [52]. Negative parts of TA at 450-570 nm and at 570-600 nm are due to ground-state bleaching (GSB) and stimulated emission, respectively, which also correspond well with the maxima of steady-state absorption and emission spectra.

|
Download:
|
| Fig. 4. Femtosecond time-resolved absorption spectra (a, c, e) and the time-absorption profiles (b, d, f) in CH2Cl2 of PDI, 2PDI-Th, and fused-2PDI-Th, respectively. | |
{kind=link}
As shown in Fig. 4c, the fsTA spectra of 2PDI-Th exhibit broad GSB band in the range of 450-585 nm. Moreover, the fsTA spectra of 2PDI-Th displays no stimulated emission, which indicates the formation of excimer. Narrow positive band was also observed with the maximum at 620 nm at early times; further, it broadened. We assign this band to the excimer absorption [42-44, 50]. The kinetics of this absorption band indicates that the rapid formation of excimer occurs within 6 ps (Fig. 4d and Table S5 in Supporting information). It is in agreement with the two-step mechanism of the excimer formation: hot excimer is formed within ultrashort time of < 100 fs; vibrational cooling transfers hot excimer into relaxed "cold" excimer within 4-8 ps [42-44, 50]. This is also accompanied by significant variation in the molecular configuration; the molecular geometry has changed a lot with a dihedral angle of 30° from S0 to S1. Kinetic fit at 620 nm results in excimer lifetime of 500 ps (Table S5), which is in agreement with the timeresolved fluorescence data. Moreover, the transient spectral features show that the decay of the excimer results in appearance of a relatively long-lived band at 475-585 nm, which corresponds to triplet excited state absorption [50, 53]. The kinetics at 490 nm contains two time components: decay with 39 ps and a rise time of 1800 ps. Because the triplet excited state absorption overlaps with the GSB, the short time component of 39 ps is due to the GSB. The time of 1800 ps represents the generation of triplet state. The fsTA spectral data are in good agreement with the fluorescence results of TCSPC.
Compared with that of 2PDI-Th, the fsTA spectra of fused-2PDITh (Fig. 4e) is very different. The transient spectra shows a broad GSB band in the region 450-520 nm, a weak stimulated emission (SE) band at 525 nm [42-44] and two positive absorption bands covering between 525 nm and 660 nm. Due to strong overlap with GSB, the ESA in the range of 480-560 nm is not detected. The absorption spectrum with peak at 610 nm is attributed to S1-Sn transition. Decay of the singlet excitons with τ =3.4 ns results in appearance of 530 nm ESA with τrise=4.7 ns (Fig. 4f and Table S6 in Supporting information), i.e., decay of singlet state results in triplet state formation via ISC.
To extract the excited state dynamics, evolution-associated spectra (EAS) of global fit are as shown in Fig. S5 (Supporting information). According to the results of global fitting, for 2PDI-Th (Fig. S5a), the EAS with the shortest time component shows that singlet excited state decay and excimer growth occur within 1.5 ps. Moreover, the decay time of excimer is 630 ps. For fused-2PDI-Th (Fig. S5b), its singlet lifetime is 4.5 ns, that is, the time of the intersystem crossing process is 4.5 ns. The results of the global fit are basically consistent with the results of the previous biexponential fit.
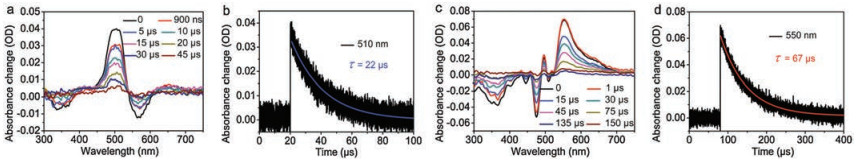
Nanosecond transient absorption (nsTA) spectroscopy was used to investigate the long-lived kinetic processes. For PDI, long-lived transient excitons were not traced in nsTA, neither in fsTA. As shown in Fig. 5a, the spectra of 2PDI-Th in dichloromethane exhibits two GSB bands in the range of 310-390 nm and 540-600 nm at λexc = 532 nm. Due to the overlap with the ESA, GSB in the wavelength range of 400–540 nm is not observed. Moreover, there is a strong ESA from 480 nm to 540 nm, which is assigned to the triplet state absorption. Through kinetic fitting at 510 nm, triplet state decays with lifetime of 22 μs (Fig. 5b). The nsTA spectroscopy of fused-2PDI-Th reveals a broad GSB covering 300-490 nm range, weak ESA at 490-500 nm and an intense absorption peak at 550 nm (Fig. 5c). Obviously, these two ESA bands belong to the same process, i.e., triplet state absorption. The decay trace monitored at 550 nm gives a long triplet state lifetime of 67 μs (Fig. 5d), which is factor of 3 times longer than that of 2PDI-Th. The long lifetime of the triplet state excitons is favorable to the exciton transporting and provides more possibilities for the charge separation at the donor-acceptor interface of the active layer in OSCs.

|
Download:
|
| Fig. 5. Nanosecond transient absorption spectra (a, c) and their decay trances (b, d) of 2PDI-Th and fused-2PDI-Th, respectively. | |
{kind=link}
Since both 2PDI-Th and fused-2PDI-Th own long lifetime of the triplet state excitons, we used them as the triplet photosensitizers to oxidize triplet oxygen and measured the quantum yields (QY) of singlet oxygen generation [25, 54, 55]. Using 1, 3-diphenylisobenzofuran (DPBF) as a singlet oxygen scavenger and Ru(bpy)3(PF6)2 as the standard (QY: 57.0% in acetonitrile). In non-polar solvent of hexane, a moderate value of 30.0% was obtained for fused-2PDI-Th, while a very low value of 1.3% was observed for 2PDI-Th. In the polar solvent of dichloromethane, fused-2PDI-Th gives an enhanced QY of 48.4%, however in 2PDI-Th QY is very small (4.9%). From TD-DFT calculations, the first triplet energy levels (T1) are estimated of about -1.6 and -1.0 eV for fused-2PDI-Th and 2PDITh, respectively. As we know, the T1 of singlet oxygen is at -0.98 eV, which means that the triplet excitons of 2PDI-Th and fused-2PDI-Th can sensitize the triplet oxygen. All these results indicate that fused-2PDI-Th exhibits efficient ISC and longer lifetimes of triplet excitons, which proved their important role for high efficiency OSCs.
In summary, we have performed detailed study of the excited states relaxation properties of two thiophene-coupled PDI dyads 2PDI-Th and fused-2PDI-Th. From the DFT calculations, 2PDI-Th exhibits a similar H-typed molecular configuration with a dihedral angle of 72° in the optimized ground state between the two PDI units. Especially in excited state, the dihedral angle decreases to a less value of 42°, which is beneficial for the formation of excimers. Compound fused-2PDI-Th shows an X-shaped configuration that retains the same structure from S0 to S1. Compared to that of fused-2PDI-Th, the steady-state emission spectrum of 2PDI-Th indicates weak broad fluorescence of excimers. The lifetime of the excimers (840 ps) is much shorter than those of the excitons of PDI (5.5 ns) and fused-2PDI-Th (3.3 ns). From the fsTA spectra of 2PDI-Th, the excimers are generated within 1.5 ps and decay with formation of triplet state within 0.5–1.1 ns. For fused-2PDI-Th, the triplet state is generated directly from the singlet state within 4.7 ns. The long triplet lifetime (67 μs) of fused-2PDI-Th is factor of three longer than that (22 μs) of 2PDI-Th. Moreover, for fused-2PDI-Th, the efficiency of singlet oxygen generation (in dichloromethane) is high, indicating an enhanced ISC. These results demonstrate that ring fusion is a useful method to reduce the possibility of excimer formation and extend the exciton diffusion length, which results in overall improvement of photovoltaic devices. This strategy provides not only a new insight for the high PCEs, but also a rational design for the organic optoelectronic materials to achieve high efficiency OSCs.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 21421005, 21576040, 21776037 and 21875027), the Fundamental Research Funds for the Central Universities (No. DUT19LK05), and Supercomputing Center of Dalian University of Technology.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.018.
| [1] |
A. Kamkaew, S.H. Lim, H.B. Lee, et al., Chem. Soc. Rev. 42 (2013) 77-88. DOI:10.1039/C2CS35216H |
| [2] |
J. Zhao, W. Wu, J. Sun, S. Guo, Chem. Soc. Rev. 42 (2013) 5323-5351. DOI:10.1039/c3cs35531d |
| [3] |
Z. Zhou, J. Song, L. Nie, X. Chen, Chem. Soc. Rev. 45 (2016) 6597-6626. DOI:10.1039/C6CS00271D |
| [4] |
D. Ravelli, M. Fagnoni, A. Albini, Chem. Soc. Rev. 42 (2013) 97-113. DOI:10.1039/C2CS35250H |
| [5] |
C.K. Prier, D.A. Rankic, D.W.C. MacMillan, Chem. Rev. 113 (2013) 5322-5363. DOI:10.1021/cr300503r |
| [6] |
P. Gandeepan, T. Müller, D. Zell, et al., Chem. Rev. 119 (2019) 2192-2452. |
| [7] |
J. Zhou, Z. Liu, F. Li, Chem. Soc. Rev. 41 (2012) 1323-1349. DOI:10.1039/C1CS15187H |
| [8] |
G. Chen, H. Qiu, P.N. Prasad, X. Chen, Chem. Rev. 114 (2014) 5161-5214. DOI:10.1021/cr400425h |
| [9] |
N. Yanai, N. Kimizuka, Acc. Chem. Res. 50 (2017) 2487-2495. DOI:10.1021/acs.accounts.7b00235 |
| [10] |
Y. Sun, N.C. Giebink, H. Kanno, et al., Nature 440 (2006) 908-912. DOI:10.1038/nature04645 |
| [11] |
H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature 492 (2012) 234-238. DOI:10.1038/nature11687 |
| [12] |
S.D. Stranks, G.E. Eperon, G. Grancini, et al., Science 342 (2013) 341-344. DOI:10.1126/science.1243982 |
| [13] |
M. Einzinger, T. Wu, J.F. Kompalla, et al., Nature 571 (2019) 90-94. DOI:10.1038/s41586-019-1339-4 |
| [14] |
S.R. Yost, E. Hontz, S. Yeganeh, Van Voorhis T., J. Phys. Chem. C 116 (2012) 17369-17377. DOI:10.1021/jp304433t |
| [15] |
B. Chen, Z.J. Yu, S. Manzoor, et al., Joule 4 (2020) 850-864. DOI:10.1016/j.joule.2020.01.008 |
| [16] |
N.J. Turro, J.C. Scaiano, University Science Books Sausalito, CA (2009). |
| [17] |
Y. You, W. Nam, Chem. Soc. Rev. 41 (2012) 7061-7084. DOI:10.1039/c2cs35171d |
| [18] |
Z. Liu, W. He, Z. Guo, Chem. Soc. Rev. 42 (2013) 1568-1600. DOI:10.1039/c2cs35363f |
| [19] |
R. Ziessel, B.D. Allen, D.B. Rewinska, A. Harriman, Chem. Eur. J. 15 (2009) 7382-7393. DOI:10.1002/chem.200900440 |
| [20] |
J.Z. Zhao, K.J. Xu, W.B. Yang, Z.J. Wang, F.F. Zhong, Chem. Soc. Rev. 44 (2015) 8904-8939. DOI:10.1039/C5CS00364D |
| [21] |
E. Okutan, Kamanina (Eds.) N., Fullerenes and Relative Materials, IntechOpen, London pp (2018) 127-143. |
| [22] |
J.W. Verhoeven, H.J. Ramesdonk, M.M. Groeneveld, A.C. Benniston, A. Harriman, ChemPhysChem 6 (2005) 2251-2260. DOI:10.1002/cphc.200500029 |
| [23] |
K. Ohkubo, S. Fukuzumi, Bull. Chem. Soc. Jpn. 82 (2009) 303-315. DOI:10.1246/bcsj.82.303 |
| [24] |
S. Suzuki, M. Kozaki, K. Nozaki, K. Okada, J. Photochem. Photobiol. C:Photochem. Rev. 12 (2011) 269-292. DOI:10.1016/j.jphotochemrev.2011.10.001 |
| [25] |
Y. Wu, Y. Zhen, Y. Ma, et al., J. Phys. Chem. Lett. 1 (2010) 2499-2502. DOI:10.1021/jz1008328 |
| [26] |
C. Li, H. Wonneberger, Adv. Mater. 24 (2012) 613-636. DOI:10.1002/adma.201104447 |
| [27] |
W. Jiang, Y. Li, Z. Wang, Acc. Chem. Res. 47 (2014) 3135-3147. DOI:10.1021/ar500240e |
| [28] |
F. Würthner, Saha-Möller C.R., B. Fimmel, et al., Chem. Rev. 116 (2016) 962-1052. DOI:10.1021/acs.chemrev.5b00188 |
| [29] |
P. Cheng, G. Li, X. Zhan, Y. Yang, Nat. Photonics 12 (2018) 131-142. DOI:10.1038/s41566-018-0104-9 |
| [30] |
J. Ho, O. Inganäs, R.H. Friend, F. Gao, Nat. Mater. 17 (2018) 119-128. DOI:10.1038/nmat5063 |
| [31] |
I.A. Howard, F. Laquai, P.E. Keivanidis, R.H. Friend, N.C. Greenham, J. Phys. Chem. C 113 (2009) 21225-21232. DOI:10.1021/jp907633g |
| [32] |
X. Zhan, Z. Tan, B. Domercq, et al., J. Am. Chem. Soc. 129 (2007) 7246-7247. DOI:10.1021/ja071760d |
| [33] |
D. Meng, H. Fu, C. Xiao, et al., J. Am. Chem. Soc. 138 (2016) 10184-10190. DOI:10.1021/jacs.6b04368 |
| [34] |
J. Zhang, Y. Li, J. Huang, H. Hu, et al., J. Am. Chem. Soc. 139 (2017) 16092-16095. DOI:10.1021/jacs.7b09998 |
| [35] |
Z. Cai, D. Zhao, V. Sharapov, et al., ACS Appl. Mater. Interfaces 10 (2018) 13528-13533. DOI:10.1021/acsami.8b01308 |
| [36] |
H. Wang, L. Chen, Y. Xiao, J. Mater. Chem. A 5 (2017) 22288-22296. DOI:10.1039/C7TA06804B |
| [37] |
H. Wang, J. Wu, Y. Zhang, et al., Org. Electron. 83 (2020) 105732. DOI:10.1016/j.orgel.2020.105732 |
| [38] |
H. Zhong, C.H. Wu, C.Z. Li, et al., Adv. Mater. 28 (2016) 951-858. DOI:10.1002/adma.201504120 |
| [39] |
P.E. Hartnett, H.S.S.R. Matte, N.D. Eastham, et al., Chem. Sci. 7 (2016) 3543-3555. DOI:10.1039/C5SC04956C |
| [40] |
H. Wang, L. Chen, Y. Xiao, J. Mater. Chem. C 7 (2019) 835-842. DOI:10.1039/C8TC04929G |
| [41] |
R. Katoh, E. Katoh, N. Nakashima, M. Yuuki, M. Kotani, J. Phys. Chem. A 101 (1997) 7725-7728. DOI:10.1021/jp964075u |
| [42] |
R.E. Cook, B.T. Phelan, R.J. Kamire, et al., J. Phys. Chem. A 121 (2017) 1607-1615. DOI:10.1021/acs.jpca.6b12644 |
| [43] |
K.E. Brown, W.A. Salamant, L.E. Shoer, R.M. Young, M.R. Wasielewski, J. Phys. Chem. Lett. 5 (2014) 2588-2593. DOI:10.1021/jz5011797 |
| [44] |
E.A. Margulies, L.E. Shoer, S.W. Eaton, M.R. Wasielewski, Phys. Chem. Chem. Phys. 16 (2014) 23735-23742. DOI:10.1039/C4CP03107E |
| [45] |
R.F. Fink, J. Seibt, V. Engel, et al., J. Am. Chem. Soc. 130 (2008) 12858-12859. DOI:10.1021/ja804331b |
| [46] |
A. Schubert, V. Settels, W. Liu, et al., J. Phys. Chem. Lett. 4 (2013) 792-796. DOI:10.1021/jz4000752 |
| [47] |
S. Rajaram, P.B. Armstrong, B.J. Kim, J.M.J. Fréchet, Chem. Mater. 21 (2009) 1775-1777. DOI:10.1021/cm900911x |
| [48] |
F. Würthner, Chem. Commun. (2004) 1564-1579. |
| [49] |
S.C. Lo, P.L. Burn, Chem. Rev. 107 (2007) 1097-1116. DOI:10.1021/cr050136l |
| [50] |
K.M. Lefler, K.E. Brown, W.A. Salamant, et al., J. Phys. Chem. A 117 (2013) 10333-10345. DOI:10.1021/jp4083008 |
| [51] |
J. Mei, N.L.C. Leung, R.T.K. Kwok, J.W.Y. Lam, B.Z. Tang, Chem. Rev. 115 (2015) 11718-11940. DOI:10.1021/acs.chemrev.5b00263 |
| [52] |
K.M. Felter, R.K. Dubey, F.C. Grozema, J. Chem. Phys. 151 (2019) 094301. DOI:10.1063/1.5110306 |
| [53] |
Z. Yu, Y. Wu, Q. Peng, et al., Chem. Eur. J. 22 (2016) 4717-4722. DOI:10.1002/chem.201600300 |
| [54] |
Y. Usui, Chem. Lett. 2 (1973) 743-744. DOI:10.1246/cl.1973.743 |
| [55] |
Y. Zhao, A.A. Sukhanov, R. Duan, et al., J. Phys. Chem. C 123 (2019) 18270-18282. DOI:10.1021/acs.jpcc.9b04896 |