 2020, Vol. 31
2020, Vol. 31 


Ambient organic peroxy (RO2) radical is almost the only highly reactive intermediate produced during the oxidation processes of hydrocarbons [1, 2]. As a prominent media, RO2 radical is closely related to the removal of primary pollutants and the generation of secondary pollutants. Together with hydroperoxy (HO2) and hydroxyl (OH) radicals, RO2 radicals determine the atmospheric oxidation capacity, and therefore significantly influence the formation of secondary pollutants, including tropospheric ozone (O3), organic nitrates, organic acids, and particulate matters [3-7].
The atmospheric abundance and distribution of RO2 radicals are crucial for a better understanding of tropospheric chemical oxidation [8]. The quantitative detection of RO2 is not only a critical data foundation but a useful parameter in the chemical model as well [8, 9]. However, since RO2 is highly reactive with trace gases and surfaces, similar to the other free radicals, a sensitive and precise detection system is required to measure ambient RO2, which is technically challenging to achieve.
The first field measurement approach of RO2 radicals was put forward in 1978 [10], known as the matric isolation and electron spin resonance (MIESR). It is a direct measurement technique and can distinguish different RO2 species. However, complex operation and harsh sample storage conditions limit the application of this technique [10-12]. The peroxy radical chemical amplifier (PERCA), first proposed in 1984 [13], is one of the most widely used methods due to its high operability, but it can hardly separate different RO2 species [13-17]. The same problem happens to the peroxy radical chemical ionization mass spectrometry (PerCIMS (or ROxMAS) technique, although it has higher accuracy and shorter chain length requirement [9]. The laser-induced fluorescence (LIF) is a classic technique of measuring OH and HO2 [18, 19]. Some progress has been made in measuring RO2 radicals by combining LIF with a chemical flow tube reactor [20], but only a few field observations of RO2 radicals have been reported before [21, 22].
In this work, a flow tube reactor was developed based on the previous works [20, 23] and was applied together with the Peking University LIF instrument, implementing the measurement of ambient RO2 radicals. By utilizing the active chemical priorities of some types of RO2 (long-chain alkane-, alkene- and aromatic hydrocarbon-derived) radicals (known as RO2#) [21], speciated detection of these types of RO2 was achieved.
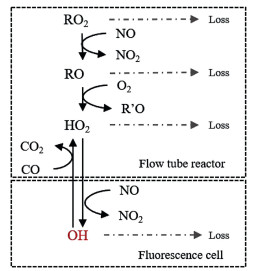
The measurement of ambient RO2 by LIF technique involves two steps of chemical conversion: a) ROx (= RO2 + HO2 + RO + OH) radicals are converted into HO2 radicals inside the flow tube reactor under a low-pressure condition; b) HO2 radicals obtained at the exit of the flow tube are further converted into OH radicals in the fluorescence cell, whereOH can be detected directly, under an even lower pressure condition. These processes are illustrated in Fig. 1.

|
Download:
|
| Fig. 1. Chemical reactions in the reactor and fluorescence cell. "Loss" in grey includes the process of wall collision and intermolecular recombination reaction; OH in red is the signal detected by the LIF directly. | |
{kind=link}
Inside the flow tube reactor, nitric oxide (NO) and carbon monoxide (CO) are introduced as reagent gases. With excessive NO, RO2 can quickly convert to organic oxy radicals (RO), followed by the formation of HO2 and carbonyl compounds (R'O) with the presence of oxygen (O2) in sampled air (Reactions 1 and 2). Generated HO2, as well as the initially existed HO2 in sampled air, can further react with NO, resulting in the generation of OH radicals (Reaction 3):
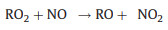
|
(1) |

|
(2) |
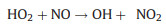
|
(3) |
As the chemical reactivity of OH is much higher than that of HO2, the prolonged residence of OH radical in the flow tube may cause significant radical loss. Excessive CO is simultaneously injected into the reactor, thereby the chemical equilibrium between OH and HO2 can be shifted to the HO2 side (Reactions 4 and 5), minimizing the radical loss in the flow tube:
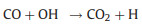
|
(4) |
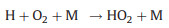
|
(5) |
Nevertheless, the conversion efficiency of RO2 to HO2 is still inevitably limited due to several radical loss processes. Firstly, all the ROx radicals can undergo termolecular recombination reactions with NO (Reactions 6-9):
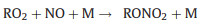
|
(6) |
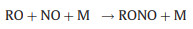
|
(7) |
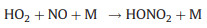
|
(8) |
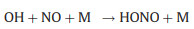
|
(9) |
Secondly, reactive collisions of ROx radicals with the inner surface of the reactor are also major loss processes need to be considered (Reactions 10–13):
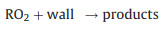
|
(10) |

|
(11) |
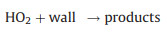
|
(12) |
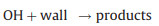
|
(13) |
Reactions with NO (Reactions 6–9) are pressure-dependent [20], so they can be suppressed effectively by lowering the pressure in the reactor. Based on the rate constants recommended by previous studies (Table S1 in Supporting information), recombination reactions become insignificant under the low-pressure condition. But wall reactions are still dominant with a reduction of total HO2 yield of ~20% [20], even after coating the inner surface of the reactor with the Teflon layer.
The central part of the airflow is collected into the fluorescence cell at the exit of the reactor. Additional NO is injected into this unit to shift the chemical equilibrium between HO2 and OH back to the OH side, which can be detected by the LIF directly. A detailed description of the detection principle can be found in previous papers [18, 19]. In brief, OH radicals can be excited by narrow-bandwidth UV laser (λ =308 nm) on a single rovibronic transition, and therefore fluoresce. Gated photon counting is used to produce a time delay to discriminate the emitted fluorescence by OH and laser stray light. The background signal is determined by turning the laser resonance on and off periodically. The conversion between fluorescence signals and radical concentrations requires a calibration.
When NO and CO are added as reagent gases into the reactor, all the ROx radicals are converted and detected in the fluorescence cell, so this measurement mode is called "ROx mode". When only CO is added, RO2 and RO cannot be converted into HO2 theoretically, so this mode was called "HOx mode" in previous studies [20]. However, it has been found afterward that for longer chain (C > 3) alkane-, alkene- and aromatic hydrocarbon-derived RO2 radicals (RO2#), they can rapidly convert to HO2 and then OH in the fluorescence cell when the concentration of NO reaches a certain level in the fluorescence cell [23, 24]. This characteristic may elevate the signal of HO2 radicals as an "RO2 interference", yet it is also a potentiality to measure different RO2 species by rising NO concentration in the fluorescence cell. In consequence, when only CO is introduced, RO2#, RO, OH, and HO2 are measured (named as "RO2# mode"). Because the concentration of RO is negligible compared to that of RO2 and HO2, so RO2 and RO2# radical concentrations can then be calculated by simultaneously measured OH and HO2 data.
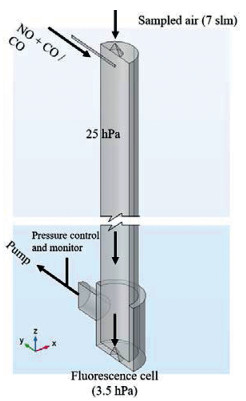
Based on the measurement principle mentioned above, the flow tube reactor was designed, as can be seen in Fig. 2. The ambient air was sampled into the reactor through a 1 mm orifice (nickel, 70°, Beam Dynamics, Inc.) with a flow rate of 7 slm (standard liters per minute at 1 atm, 20 ℃). Reagent gases, NO and CO, were immediately injected into the sampled air through a glass tube (λ = 100 mm, i.d. = 4 mm) after the inlet nozzle, attaining ultimate mixing ratios of 0.7 ppmv and 0.17% respectively. These concentrations were defined based on the simulation results of MCM 3.2 ([20]). The flow tube (λ = 830 mm, i.d. = 66 mm) is made of quartz, and its inner surface is coated with the Teflon layer. The reactor was operated at ~25 hPa. The pressure was controlled by a butterfly valve and monitored by a vacuum gauge (Edwards, APG100). Excess air in the reactor was removed by an oil-free vacuum pump (Edwards, XDS35i).

|
Download:
|
| Fig. 2. Schematic diagram of the flow tube reactor (partially broken). | |
{kind=link}
At the end of the flow tube, another nozzle (4 mm orifice, stainless steel, 70°) was used to sample the central part of the reacted gas to the fluorescence cell, in which the pressure was kept at ~4 hPa. Pure NO (Linde AG, 99.5%) was used to convert HO2 to OH, so that both RO2 and RO2# can be detected in this work.
All the reagent gases were controlled by mass flow controllers (HORIBA METRON, S48 300/HMT). Electromagnetic valves and time relays were used to achieve periodically (5 min) switch between the two measurement modes. Thus, continuous online measurement with alternating modes has a time resolution of 10 min for both RO2 and RO2#.
Based on previous studies, equal concentrations of OH and HO2 can be generated by the photolysis (λ =185 nm) of H2O at 1 atm [19, 25]. It has also been proven that equal amounts of HO2 and RO2 can be produced when the suitable hydrocarbon is mixed with humid air [8, 20, 26], as long as the introduced hydrocarbon is stabilized under 185 nm ultraviolet (UV). Hence, it is feasible to calibrate the system by using a radical source designed for OH and HO2 calibration (for a detailed description of the calibration source, see reference [20]).
The calibration method depends on the approaches to obtaining OH and HO2 data. If all the RO2, HO2, and OH are measured simultaneously by one LIF instrument, the detection sensitivities can be determined in three steps by altering reagent gases in the calibration source: a) Humid air with CO; b) humid air with hydrocarbon; c) only humid air. The fluorescence photon count rate (counts/s) of each radical species can then be calculated directly. The calculation method can be found in the previous paper [20].
However, if the OH and HO2 data are obtained from another instrument, which is the case of this work, the photon count rate cannot be calculated directly. Concentrations of ROx, HO2, and OH need to be calculated before doing the subtraction. In this case, three parameters should be considered: a) Conversion efficiency of RO2 to HO2 in the reactor (r1), b) conversion efficiency of HO2 to OH in the fluorescence cell (r2), c) the sensitivity of HO2 radicals (SHO2/ [HO2]0).
Since pure NO is used in the fluorescence cell, 100% HO2 is assumed to be converted into OH (r2 = 1). The conversion efficiency of RO2 to HO2 is referenced by CH3O2, which was simulated by the Master Chemical Mechanism (MCM v3.3). The sensitivity of ROx radicals can be calculated via Eq. 1:

|
(1) |
An ambient air measurement was performed in summer 2019, at a wetland park in Chengdu, China (30.4259°N, 103.847126°E). The site is in the suburbs, surrounded by plenty of vegetation. However, because of the nearby China Civil Aviation Flight Academy, many helicopters were flying above the site during the campaign, which may lead to primary emissions of pollutants. The instrument was placed ~10 m above ground, adjacent to the other LIF (used to measure HO2 and OH).
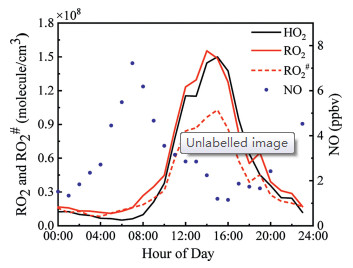
Fig. 3 shows the averaged diurnal profiles of RO2 and RO2# radicals from 26th August to 12th September. The variations of RO2 and RO2# were highly correlated with HO2 radicals. These photochemically formed radicals peaked at around 14:00 with concentrations of 1.55 × 108 (RO2), 1.04 × 108 (RO2#), and 1.50 × 108 (HO2) molecule/cm3, when the solar actinic flux reached the highest value. In the meanwhile, NO concentration reached the lowest values at noon and peaked in the early morning with a concentration of ~8 ppbv. An apparent negative correlation between peroxy radicals and NO can be observed. A small peak of RO2 and RO2# radicals was found at around 20:00, indicating the existence of radical source in the evening. A strong positive correlation between NO3 and RO2 radicals has been found in a previous study [27], implying the rising NO3 radicals may be the possible reason for this small peak at night. The concentration level and diurnal pattern of RO2 are similar to what has been reported in previous studies [20-23], besides the RO2#/RO2 (~60% in this case) was a little higher than before. The divergence between RO2# and RO2 can hardly be observed in the early morning when NO concentration was increasing rapidly. A possible reason is that the ambient NO promoted the conversion of RO2 in the converter or even in the atmospheric, invalidating the separation of RO2# radicals. Volatile organic compounds (VOCs) were also measured in this campaign (Table S2 and Fig. S1 in Supporting information). Ethene was the most abundant VOC with a proportion of 4.49% that can generate RO2#, while ethyne was the most abundant among all the VOCs. The ratio of RO2#-produced VOCs to total VOCs was about 35.5%.

|
Download:
|
| Fig. 3. The diurnal means of RO2, RO2#, HO2, and NO (1 -h average). | |
{kind=link}
The photochemical O3 production rate (P(O3)) can be calculated by the oxidation rate of NO to NO2 through its reaction with peroxy radicals (HO2 and RO2, Reactions 1–3) [28-30]. Without considering the loss processes of NO2 (e.g., the reaction of NO2 and OH or the formation of organic nitrates by RO2), the upper limit for P(O3) can be estimated by Eq. 2 [31],
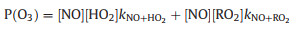
|
(2) |
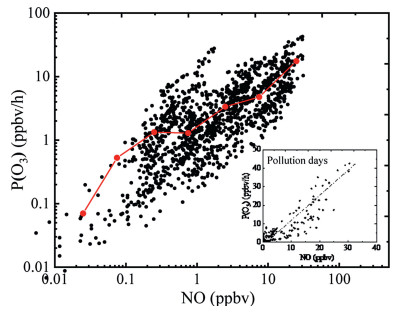
where kRO2+NO and kHO2+NO denote the rate constants of the reaction of RO2 + NO and HO2 + NO. Fig. 4 shows the dependence of P(O3) on NO. The P(O3) increased with the rising of NO concentration, except for a small decline of around 1 ppbv of NO. The correlation between NO and P(O3) in pollution days has also been reported in Fig. 4 with a relatively strong positive correlation (R2 = 0.78), indicating that the suburb Chengdu is NOx sensitive for the ozone production.

|
Download:
|
| Fig. 4. NO dependence of P(O3) and the correlation between NO and P(O3) in pollution days. | |
{kind=link}
In conclusion, a low-pressure flow tube reactor coupling with the LIF instrument was developed and successfully applied in the field measurement of RO2 and RO2#. RO2 radicals were firstly converted to HO2 radicals in the reactor and then to OH in the fluorescence cell. Longer chain (C > 3) alkane, alkene and aromatic hydrocarbon-derived RO2 radicals (RO2#) can be determined separately due to their selective chemical properties. The results of field measurements have proven that the designed reactor is an effective and convenient extension for the LIF technique. High NO concentration may affect the speciated measurement results, which limits the application of this technique in urban areas. Further studies on improving the performance of speciated measurements are still needed.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe work was supported by the National Key R&D Program of China (No. 2017YFC0209402) and the Beijing Natural Science Foundation, China (No. JQ19031). The authors gratefully acknowledge the support and assistance from Prof. Limin Zeng and Prof. Xin Li in the field campaign of Chengdu.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.07.051.
| [1] |
G.P. Brasseur, J.J. Orlando, G.S. Tyndall, Atmospheric Chemistry and Global Change. New York: Oxford University Press, 1999.
|
| [2] |
T. Berndt, W. Scholz, B. Mentler, et al., Angew. Chem. Int. Ed. 57 (2018) 3820-3824. DOI:10.1002/anie.201710989 |
| [3] |
Q. Zou, H. Song, M. Tang, K. Lu, Chin. Chem. Lett. 30 (2019) 2236-2240. DOI:10.1016/j.cclet.2019.07.041 |
| [4] |
R. Atkinson, D.L. Baulch, R.A. Cox, et al., J. Phys. Chem. Ref. Data 26 (1997) 521-1011. DOI:10.1063/1.556011 |
| [5] |
R. Atkinson, J. Arey, Chem. Rev. 103 (2003) 4605-4638. DOI:10.1021/cr0206420 |
| [6] |
R. Atkinson, J. Arey, S.M. Aschmann, Atmos. Environ. 42 (2008) 5859-5871. DOI:10.1016/j.atmosenv.2007.08.040 |
| [7] |
X.P. Yang, H.C. Wang, Z.F. Tan, K.D. Lu, Y.H. Zhang, Acta Chim. Sin. 77 (2019) 613-624. DOI:10.6023/A19030094 |
| [8] |
M. Hanke, J. Uecker, T. Reiner, F. Arnold, Int. J. Mass Spectrom. 213 (2002) 91-99. DOI:10.1016/S1387-3806(01)00548-6 |
| [9] |
T. Reiner, M. Hanke, F. Arnold, J. Geophys. Res. 102 (1997) 1311-1326. DOI:10.1029/96JD02963 |
| [10] |
D. Mihelcic, D.H. Ehhalt, G.F. Kulessa, et al., Pure Appl. Geophys. 116 (1978) 530-536. DOI:10.1007/BF01636905 |
| [11] |
D. Mihelcic, P. Musgen, P. Müsgen, D.H. Ehhalt, J. Atmos. Chem. 3 (1985) 341-361. DOI:10.1007/BF00122523 |
| [12] |
D. Mihelcic, A. Voltz-Thomas, A. Volz-Thomas, et al., J. Atmos. Chem. 11 (1990) 271-297. DOI:10.1007/BF00118353 |
| [13] |
C.A. Cantrell, D.H. Stedman, Geophys. Res. Lett. 9 (1982) 846-849. DOI:10.1029/GL009i008p00846 |
| [14] |
D.R. Hastie, M. Weissenmayer, J.P. Burrows, G.W. Harris, Anal. Chem. 63 (1991) 2048-2057. DOI:10.1021/ac00018a029 |
| [15] |
J. Hu, D.H. Stedman, Anal. Chem. 66 (1994) 3384-3393. DOI:10.1021/ac00092a015 |
| [16] |
K.C. Clemitshaw, L.J. Carpenter, S.A. Penkett, M.E. Jenkin, J. Geophys. Res. 102 (1997) 25405-25416. DOI:10.1029/97JD01902 |
| [17] |
Y. Chen, C. Yang, W. Zhao, et al., Analyst 141 (2016) 587-5878. |
| [18] |
F. Holland, M. Hessling, A. Hofzumahaus, J. Atmos. Sci. 52 (1995) 3393-3401. DOI:10.1175/1520-0469(1995)052<3393:ISMOTO>2.0.CO;2 |
| [19] |
F. Holland, J. Geophys. Res. 108 (2003) 8246. DOI:10.1029/2001JD001393 |
| [20] |
H. Fuchs, F. Holland, A. Hofzumahaus, Rev. Sci. Instrum. 79 (2008) 84104. DOI:10.1063/1.2968712 |
| [21] |
Z. Tan, H. Fuchs, K. Lu, et al., Atmos. Chem. Phys. 17 (2017) 663-690. DOI:10.5194/acp-17-663-2017 |
| [22] |
Z. Tan, F. Rohrer, K. Lu, et al., Atmos. Chem. Phys. 18 (2018) 12391-12411. DOI:10.5194/acp-18-12391-2018 |
| [23] |
L.K. Whalley, M.A. Blitz, M. Desservettaz, P.W. Seakins, D.E. Heard, Atmos. Meas. Tech. 6 (2013) 3425-3440. DOI:10.5194/amt-6-3425-2013 |
| [24] |
H. Fuchs, B. Bohn, A. Hofzumahaus, et al., Atmos. Meas. Tech. 4 (2011) 1209-1225. DOI:10.5194/amt-4-1209-2011 |
| [25] |
A. Hofzumahaus, U. Aschmutat, U. Brandenburger, et al., J. Atmos. Chem. 31 (1998) 227-246. DOI:10.1023/A:1006014707617 |
| [26] |
D.J. Creasey, P.A. Halford-Maw, D.E. Heard, M.J. Pilling, B.J. Whitaker, Faraday Trans. 93 (1997) 2907-2913. DOI:10.1039/a701469d |
| [27] |
A. Geyer, K. Bächmann, A. Hofzumahaus, et al., J. Geophys. Res. 108 (2003) 8249. DOI:10.1029/2001JD000656 |
| [28] |
A. Volz, D. Mihelcic, P. Müsgen, et al., Tropospheric Ozone: Regional and Global Scale Interactions, Springer, Netherlands, Dordrecht, 1988.
|
| [29] |
B.A. Ridley, J. Geophys. Res. 97 (1992) 10-388. |
| [30] |
L. Kleinman, Y.N. Lee, S.R. Springston, et al., J. Geophys. Res. 100 (1995) 7263-7273. DOI:10.1029/95JD00215 |
| [31] |
D. Mihelcic, F. Holland, A. Hofzumahaus, et al., J. Geophys. Res. 108 (2003) 8254. DOI:10.1029/2001JD001014 |