 2020, Vol. 31
2020, Vol. 31 


b Key Laboratory of Water and Wastewater Treatment(HUST), MOHURD, Wuhan 430074, China;
c Three Gorges Base Development Co. Ltd., Yichang 443002, China
Phenols are the cyclic compounds containing the aromatic ring with the phenyl hydroxyl, which are closely associated with the human production and life. On one hand, they are the basic raw materials for organic chemical industry, for instance, phenol and bisphenol A are the basic materials for phenolic resins, an important matrix in fiber-reinforced composites to prepare materials with low fire-smoke toxicity [1]. On the other hand, phenols are also important pollutants ubiquitous in the natural environment, and eleven phenols were classified as the priority pollutants by US Environmental Protection Agency (USEPA) [2]. Phenols are highly toxic even at low concentration, they are also resistant to natural degradation and therefore persist in the environment [3], thus, it is essential to develop novel physicalchemical technologies of high efficiency for phenols treatment [4].
Among the various treatment methods, advanced oxidation processes (AOPs) through the generation of reactive oxygen species (ROS), i.e., sulfate radical (SO4·-, E0 = 2.52–3.08 V vs.NHE) and hydroxyl radical (·OH, E0 = 1.89–2.72 V vs. NHE) [5, 6], have been widely applied to non-selectively decompose numerous biorefractory organics [7]. Peroxydisulfate (PDS), peroxymonosulfate (PMS) and hydrogen peroxide (H2O2) are the most studied oxidants to produce SO4·- and ·OH [8, 9], while the high chemical, storage and transportation costs limit their applications in real wastewater treatment. Oxygen (O2) is the second most abundant gas in the atmosphere, and it also the greenest and the most economic oxidant to produce ·OH. However, the O2 activation is rather difficult due to its low redox potential (E0 = -0.33 V) [10]. From the aspect of thermodynamics, the Arrhenius activation energy for the activation of O2 by Fe2+ was determined to be 51 kJ/mol [11], which was larger than the value of the Fenton reaction (Fe2+ + H2O2, 38.18 kJ/mol [12]). From the aspect of dynamics, the second order reaction rate constant for Fe2+ and O2 was five orders of magnitude lower than of Fe2+ and H2O2 [13]. Thus, additional energy input, i.e., ultraviolet and electricity, are usually needed for the efficient activation of O2 [14-16], but these processes met a series of problems in the practical use such as high treatment cost and high requirement of water quality. Thus, achieving the facile and efficient activation of O2 has become a pressing problem.
Copper oxide (CuO) was proved to be an efficient Fenton-like catalyst for H2O2 and persulfate activation [17, 18], and it also showed high efficiency for O2 activation through the surface oxygen vacancies, which were widely used for antimicrobial applications through the generation of common ROS, i.e., superoxide anions (O2·-) [19]. Obviously, the amount of generated ROS depended on the quantity and reactivity of the CuO surface oxygen vacancies, which was reported to be further determined by the dimension and shape of the CuO particles [19, 20]. For instance, the surface area of nano-CuO was much higher than that of microCuO, and Applerot et al. [19] reported that more radicals were produced as the particle size of CuO become smaller. Meanwhile, Gilbertson et al. [20] recently showed that CuO nanosheets possessed much higher surface reactivity and antimicrobial activity than CuO nanoparticles (sphere-like shape) and bulk CuO. Overall, the catalytic ability of CuO was greatly influenced by its dimension and shape, but the underlying mechanism was still unknown. Our previous study has demonstrated that hydroxylamine (HA) could accelerate the Cu2+/Cu+ cycle, thus greatly enhanced the O2 activation by Cu nanoparticles (nCu) to generate Cu(Ⅲ) for diclofenac (DCF) degradation, while the potential toxicity of HA could bring addition risks on the environment. For the first time, we demonstrated that O2 could be efficiently activated by the surface Cu+ of CuO nanoparticles for ultrafast 2, 4-dichlorophenol (2, 4-DCP) degradation without the addition of any external chemical, but how the particle size affects the surface properties and the catalytic ability of CuO needed to be further clarified.
Therefore, in this study, four CuO samples with different particle size were selected as the research object, the effect of the dimension of CuO on 2, 4-DCP degradation and the involved ROS were firstly studied, and then the size-dependent quantity and reactivity of CuO surface reaction sites, as well as their relationship with the 2, 4-DCP degradation ratio were comprehensive studied. The results provided valuable insight into the development of sizeproperty-function parametric relationships for CuO.
All chemicals and analytical methods were provided in Text S1 (Supporting information). Batch degradation experiments were conducted in a 100 mL beaker, the initial concentration of CuO and 2, 4-DCP were set as 2 g/L and 0.012 mmol/L, respectively, and the initial pH was adjusted to 6.0 by using 0.5 mmol/L NaOH/H2SO4. A thermostat circulator was used to control the reaction temperature at 25 ℃. Water samples were withdrawn at predetermined intervals and analyzed immediately after filtration through 0.22 μm membrane. All degradation experiments were conducted at duplicates.
The size distribution diagrams presented in Fig. S1 (Supporting information) showed that the dimension of the four CuO samples were 40–80, 60–100, 35–65 and 400-700 nm, respectively, they were named as CuO-80, CuO-100, CuO-60 and CuO-600, respectively. Besides, the crystal structures of the four CuO samples were characterized by high-resolution transmission electron microscopy (HR-TEM) (Fig. S2 in Supporting information). As shown in Fig. S2c, the Fast Fourier transform (FFT) analysis showed that CuO-80 contained (200), (-112) and (110) crystal planes, while they were (200), (110) and (002) for CuO-100, (110) for CuO-60 and (110) and (111) for CuO-600 in Figs. S2g, k and o, respectively. X-ray diffraction (XRD) characterizations were then conducted to demonstrate the crystal structures of the four CuO samples, the results (Fig. S3 in Supporting information) showed that all the four CuO samples exhibited the diffraction pattern of monoclinic CuO (JCPDS No. 48-1548) [21], with well-crystalline and high purity crystal phases. The facets in the XRD patterns were in accordance with the observed facets in HR-TEM spectra. It was reported that the change in the growth direction and morphology of nanostructures could alter the intensity of the XRD peaks [22]. Among the four CuO samples, significant changes were observed in the intensity of two major peaks at 2θ values of 35.5° and 38.7°, with respect to (-111/002) and (111/200) plane. The corresponding intensity ratios of the two peaks were calculated as 1.09, 1.12, 1.08 and 1.01 for CuO-80, CuO-100, CuO-60 and CuO-600, respectively. This indicated that the percentages of the {001} facets for nanoCuO were much higher than that of micro-CuO. It would be benefit for the adsorption and activation of O2 occurring at the surface of nano-CuO, since all exposed atoms on {001} facets of CuO were Cu [23]. The N2 adsorption-desorption isotherms of the four CuO samples were shown in Fig. S4 (Supporting information). The amount of surface reaction sites was highly depended on the Brunauer-Emmet-Teller (BET) surface area of the catalysts, and it was determined to be 10.9, 4.1, 4.9 and 1.1 m2/g for CuO-80, CuO-100, CuO-60 and CuO-600, respectively. The above results showed that the quantity and reactivity of surface reaction sites of CuO was totally different, and both of them could affect the catalytic ability of CuO for O2 activation.
Electron paramagnetic resonance (EPR) experiments were firstly conducted to study the reactive species involved for 2, 4-DCP degradation in the CuO/O2 systems, the results were shown in Fig. 1. Fig. 1a displayed that a quartet peak and a sextet peak of signals were observed in the four CuO/O2 systems by using 5, 5-dimethyl-1-pyrroline N-oxide (DMPO) as the trapping agent, the two series of peaks could be recognized as DMPO-OH and DMPOCH3 adducts, representing the generation of ·OH and Cu3+, respectively [24, 25]. When 2, 2, 6, 6-tetramethyl-4-piperidone (TEMP) was used as the trapping agent, a triplet peak signal of TEMP-1O2 emerged (Fig. 1b), indicating the presence of singlet oxygen (1O2) [26]. In addition, since O2·- is very unstable and could undergo facile disproportionation rather than slow reaction with DMPO in aqueous solution [27], the trapping of O2·- was conducted in methanol media, and the detection of DMPO-OOH adduct of characteristic six peaks indicated the presence of O2·- (Fig. 1c). The above results indicated that ·OH, Cu3+, 1O2 and O2·- were generated in the CuO/O2 systems, the reaction mechanism was thus proposed. Since certain amount of oxygen vacancies and Cu+ exist on the surface of CuO [28, 29], O2 was firstly activated by surface Cu+ to generate O2·- (Eq. 1), and then a series of radical reactions were triggered based on the heterogeneous-homogeneous Cu+/Cu2+ cycle as described in Eqs. 2–5.
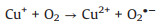
|
(1) |

|
(2) |
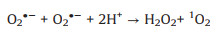
|
(3) |
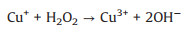
|
(4) |

|
(5) |

|
Download:
|
| Fig. 1. The EPR signals of the (a) DMPO-CH3 and DMPO-OH (aqueous solution), (b) TEMP-1O2 (aqueous solution), (c) DMPO-OOH (methanol media) adducts and (d) the relationship between the intensity of DMPO-OOH and the removal ratio of 2, 4-DCP in the four CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600. Conditions: [CuO] = 2 g/L, [DMPO] = 50 mmol/L, pH 6.0 and 25 ℃. | |
{kind=link}
Besides, as shown in Fig. 1d, the intensity of DMPO-OOH was highly correlated with the removal ratio of 2, 4-DCP, indicating the main reactive species for 2, 4-DCP degradation in the CuO/O2 systems was O2·-/HO2·. This result was further confirmed by the scavenging experiments in the CuO-80/O2 system (Fig. S5 in Supporting information). Furfuryl alcohol (FFA) was used to scavenge 1O2 [30], benzoquinone (BQ) was used for O2·- [30] and ethanol (EA) was used for ·OH and Cu3+ [31]. It can be seen that FFA and EA have no effect on 2, 4-DCP degradation, while BQ could significantly suppress the 2, 4-DCP degradation in the CuO-80/O2 system. This indicated that O2·- would be the predominant reactive species for 2, 4-DCP degradation, which was consistent with the EPR results. It also noted that the reactive species generated in the CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600 were totally identical, indicating the catalytic mechanism of O2 by CuO was independent of the dimension of CuO. The different catalytic abilities of the four CuO samples were ascribed to their different quantity and reactivity of the surface reaction sites (surface Cu+ and O2).
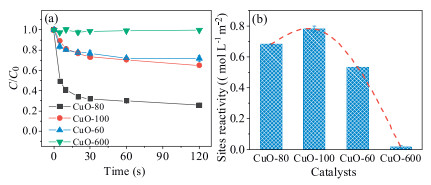
The degradation profiles of 2, 4-DCP in the CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600 were shown in Fig. 2a. It can be seen that nano-CuO could activate O2 at an ultrafast rate, and the 2, 4-DCP degradation processes were almost complete within 30 s. The 2, 4-DCP removal amount followed the order of CuO-80 > CuO-100 > CuO-60>> CuO-600, implying more ROS was generated as the particle size decreasing, which might ascribe to the larger quantity and higher reactivity of the reaction sites for nano-CuO. The sites reactivity of the four CuO samples, defined as the surface area normalized 2, 4-DCP removal amount within 120 s, were therefore calculated as shown in Fig. 2b. The sites reactivity of CuO generally followed the order of CuO-100 > CuO-80 > CuO-60>> CuO-600. It indicated that the surface reaction sites of nano-CuO was far reactive than micro-CuO.However, as the dimension of CuO less than 100 nm, the CuO sites reactivity was independently of its particle size, as described above, the exposed facets could also affect the reactivity of CuO.

|
Download:
|
| Fig. 2. (a) The degradation profiles of 2, 4-DCP in the CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600, (b) the surface sites reactivity of the four CuO samples. Conditions: [CuO] = 2 g/L, [2, 4-DCP] = 0.012 mmol/L, pH 6.0 and 25 ℃. | |
{kind=link}
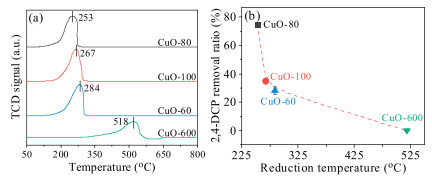
H2 temperature programmed reduction (H2-TPR) experiments were then carried out to study the reactivity of the CuO reaction sites, Fig. 3a showed that the reduction peak of CuO-80, CuO-100, CuO-60 and CuO-600 centered at 253, 267, 284 and 518 ℃, respectively, indicated that nano-CuO was more reactive than micro-CuO, which was accordance with the experimental results. The higher reactivity of nano-CuO can be ascribed to the abundant surface unsaturated coordination sites originated from oxygen vacancy on the surface of nano-CuO, which showed a higher efficiency for adsorption and dissociation of H2 during the reduction process [32]. The reactive oxygen vacancy also showed superior adsorption affinity for O2, the adsorption energy of O2 by oxygen-deficient CuO(111) surface was greater than that of perfect CuO(111) surface from an aspect of density functional theory (DFT) calculation [33]. The adsorbed O2 was weakly charged, and the reactivity of which was reported to be 4 orders of magnitude higher than that of lattice CuO oxygen [28]. As can be seen in Fig. 3b, more 2, 4-DCP was removed as the reduction temperature decreasing, further confirmed that the reactivity of surface sites determined the 2, 4-DCP removal amount to a large extent. It also noted that the reactivity of CuO-80 was higher than that of CuO-100 as shown in Fig. 3a, while opposite result was obtained from the view of surface area normalized 2, 4-DCP removal amount (Fig. 2b), this could be ascribed to the higher proportion of ROS was self-quenched since the generation of ROS was too fast as in the case of CuO-80.

|
Download:
|
| Fig. 3. H2-TPR profiles of CuO-80, CuO-100, CuO-60 and CuO-600 (a, b). | |
{kind=link}
On the basis of the above analysis, the quantity of the surface reaction sites could also affect the removal ratio of 2, 4-DCP in the CuO/O2 system. It has been reported that the surface oxygen vacancy showed superior affinity for O2 and served as the O2 binding sites on the surface of CuO [32, 33], thus, the amount of surface O2 could be revealed by the quantity of surface Cu+ or surface oxygen vacancy.
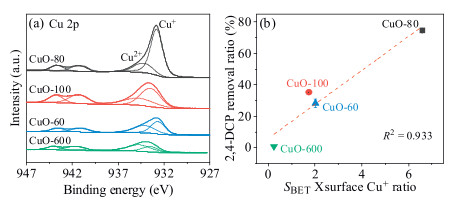
X-ray photoelectron spectroscopy (XPS) analysis was then conducted to study the quantity of Cu+ on the surface of CuO-80, CuO-100, CuO-60 and CuO-600. As depicted in Fig. 4a, the Cu 2p peak located at 934.3 eV and the corresponding satellite peaks at 938-948 eV were the XPS characteristic of Cu2+, while the peak at 932.8 eV was ascribed to Cu+ [28]. The surface Cu+ contents were then calculated to be 65.8%, 42.3%, 41.1% and 23.9% for CuO-80, CuO-100, CuO-60 and CuO-600, respectively. It was obviously that much higher proportion of Cu+ was presented on the surface of nano-CuO. The absolute amount of surface Cu+ was calculated by SBET × surface Cu+ content, it can be seen from Fig. 4b that the absolute amount of surface Cu+ was positively associated with the removal ratio of 2, 4-DCP, the correlation coefficient (R2) was 0.933, demonstrated that the quantity of CuO surface reaction sites was one of the key factors in determining the 2, 4-DCP degradation. However, as shown in Fig. 4b, the surface Cu+ quantity of CuO-60 was higher than that of CuO-100, while more 2, 4-DCP was degraded by using CuO-100, indicated that the catalytic ability of CuO depended not just on the quantity of surface reaction sites, but also their reactivity.

|
Download:
|
| Fig. 4. (a) the XPS Cu 2p spectra of CuO-80, CuO-100, CuO-60 and CuO-600, (b) the relationship between the amount of surface Cu+ and the 2, 4-DCP removal ratio in the CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600. | |
{kind=link}
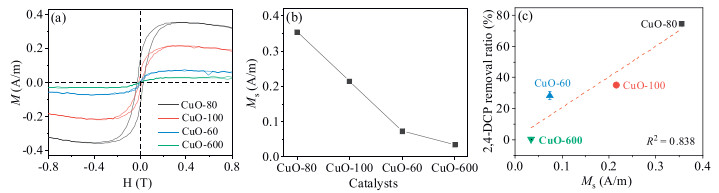
Besides, it was reported that bulk CuO show roomtemperature paramagnetism (PM), while nano-CuO presents ferromagnetism (FM), as the size of CuO decreasing, the increasing oxygen vacancy leading to the larger saturation magnetization (Ms) [34]. Fig. 5a presented the magnetization versus magnetic field ((M-H)) curves for CuO-80, CuO-100, CuO-60 and CuO-600 after paramagnetism corrections. The hysteresis loops indicated that the nano-CuO samples, i.e., CuO-80, CuO-100 and CuO-60, had visible room-temperature FM, while CuO-600 showed PM at room temperature, the Ms values of CuO-80, CuO-100, CuO-60 and CuO-600 were 0.36, 0.22, 0.073 and 0.035 A/m, respectively (Fig. 5b), indicating more oxygen vacancy, as well as more surface Cu+ and O2, were presented on the surface of nanoCuO. As shown in Fig. 5c, there still existed the positive correlation between the Ms values and the removal amount of 2, 4-DCP, while the R2 was merely 0.838, further evidenced that both the quantity and the reactivity of the CuO surface reaction sites played important roles in the fast generation of reactive species for 2, 4-DCP degradation.

|
Download:
|
| Fig. 5. (a) (M-H) curves for CuO-80, CuO-100, CuO-60 and CuO-600 after paramagnetism corrections, (b) the saturation magnetization values (Ms) of the four CuO, (c) the relationship between the Ms values and the 2, 4-DCP removal ratio in the CuO/O2 systems adopting CuO-80, CuO-100, CuO-60 and CuO-600. | |
{kind=link}
In conclusion, this study demonstrated a high-efficient AOPs based on CuO for O2 activation and 2, 4-DCP degradation. The catalytic ability of CuO was highly depended on its dimension, and nano-CuO was far reactive than micro-CuO. On one hand, higher proportion of surface reaction sites and larger BET surface area could lead to the higher quantity of reaction sites for CuO as the particle size decreasing. On the other hand, the absorbed O2 on the surface of nano-CuO was more reactive than that of micro-CuO due the presence of surface oxygen vacancy. Overall, both the quantity and the reactivity of the reaction sites (surface Cu+ and O2) on the surface of CuO were responsible for the higher catalytic ability of nano-CuO. However, the reactivity of surface reaction sites not only depended on the dimension of CuO, but also depended on crystalline factors, i.e., the exposed facets, which need further studies.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis study is financially supported by the National Natural Science Foundation of China (Nos. 21677055 and 21407052), National Key Research and Development Program of China (2019YFC1805204), Project of Three Gorges Corporation (No. JDZC-FW-20-001), and the Fundamental Research Funds for the Central Universities, HUST (Nos. 2017KFXKJC004 and 2016YXMS287). Analytic and Testing Centre of Huazhong University of Science & Technology is thanked for the advanced analytic operations.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.06.040.
| [1] |
Q. Bu, H. Lei, S. Ren, et al., Bioresour. Technol. 102 (2011) 7004-7007. DOI:10.1016/j.biortech.2011.04.025 |
| [2] |
Z. Xiong, H. Zhang, W. Zhang, B. Lai, G. Yao, Chem. Eng. J. 359 (2019) 13-31. DOI:10.1016/j.cej.2018.11.111 |
| [3] |
T. Al-Khalid, M.H. El-Naas, Crit. Rev. Environ. Sci. Technol. 42 (2012) 1631-1690. DOI:10.1080/10643389.2011.569872 |
| [4] |
X. Shen, F. Xiao, H. Zhao, et al., Environ. Sci. Technol. 54 (2020) 4564-4572. DOI:10.1021/acs.est.9b05896 |
| [5] |
M. Huang, W. Xiang, T. Zhou, et al., Sci. Total Environ. 697 (2019) 134220. DOI:10.1016/j.scitotenv.2019.134220 |
| [6] |
C. Wang, Y. Liu, T. Zhou, et al., Chin. Chem. Lett. 30 (2019) 2231-2235. DOI:10.1016/j.cclet.2019.08.055 |
| [7] |
T. Zhou, X. Zou, J. Mao, X. Wu, Appl. Catal. B:Environ. 185 (2016) 31-41. DOI:10.1016/j.apcatb.2015.12.004 |
| [8] |
Y. Li, W. Xiang, T. Zhou, M. Huang, C. Wang, et al., Chin. Chem. Lett. (2020). DOI:10.1016/j.cclet.2020.01.032 |
| [9] |
M. Ma, L. Chen, J. Zhao, W. Liu, H. Ji, Chin. Chem. Lett. 30 (2019) 2191-2195. DOI:10.1016/j.cclet.2019.09.031 |
| [10] |
Y. Nosaka, A.Y. Nosaka, Chem. Rev. 117 (2017) 11302-11336. DOI:10.1021/acs.chemrev.7b00161 |
| [11] |
R.Ž. Vracar, K.P. Cerovć, Hydrometallurgy 44 (1997) 113-124. DOI:10.1016/S0304-386X(96)00035-7 |
| [12] |
S. Karthikeyan, A. Titus, A. Gnanamani, A.B. Mandal, G. Sekaran, Desalination 281 (2011) 438-445. DOI:10.1016/j.desal.2011.08.019 |
| [13] |
A.M. Jones, P.J. Griffin, R.N. Collins, T.D. Waite, Geochim. Cosmochim. Acta 145 (2014) 1-12. DOI:10.1016/j.gca.2014.09.020 |
| [14] |
M. Huang, T. Zhou, X. Wu, J. Mao, Water Res. 119 (2017) 47-56. DOI:10.1016/j.watres.2017.03.008 |
| [15] |
H. Zhao, X. Shen, Y. Chen, et al., Chem. Commun. 55 (2019) 6173-6176. DOI:10.1039/C9CC02580D |
| [16] |
Y. Liu, M. Zhang, L. Wang, et al., Chin. Chem. Lett. 31 (2020) 805-808. DOI:10.1016/j.cclet.2019.05.045 |
| [17] |
Y. Feng, C. Liao, K. Shih, Chemosphere 154 (2016) 573-582. DOI:10.1016/j.chemosphere.2016.04.019 |
| [18] |
T. Zhang, Y. Chen, Y. Wang, et al., Environ. Sci. Technol. 48 (2014) 5868-5875. DOI:10.1021/es501218f |
| [19] |
G. Applerot, J. Lellouche, A. Lipovsky, et al., Small 8 (2012) 3326-3337. DOI:10.1002/smll.201200772 |
| [20] |
L.M. Gilbertson, E.M. Albalghiti, Z.S. Fishman, et al., Environ. Sci. Technol. 50 (2016) 3975-3984. DOI:10.1021/acs.est.5b05734 |
| [21] |
A. Ananth, S. Dharaneedharan, M.S. Heo, Y.S. Mok, Chem. Eng. J. 262 (2015) 179-188. DOI:10.1016/j.cej.2014.09.083 |
| [22] |
Y. Zhang, Y. Ji, J. Li, et al., Nano Res. 11 (2017) 804-819. |
| [23] |
X. Du, Y. Zhang, F. Si, et al., Chem. Eng. J. 356 (2019) 178-189. DOI:10.1016/j.cej.2018.08.216 |
| [24] |
L. Khachatryan, B. Dellinger, Environ. Sci. Technol. 45 (2011) 9232-9239. DOI:10.1021/es201702q |
| [25] |
M. Zhang, J. He, Y. Chen, et al., Chin. Chem. Lett. (2020). DOI:10.1016/j.cclet.2020.05.001 |
| [26] |
Z. Yang, J. Qian, A. Yu, B. Pan, Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 6659-6664. DOI:10.1073/pnas.1819382116 |
| [27] |
J. Liao, K. Li, H. Ma, et al., Chin. Chem. Lett. (2020). DOI:10.1016/j.cclet.2020.03.081 |
| [28] |
D.A. Svintsitskiy, T.Y. Kardash, O.A. Stonkus, et al., J. Phys. Chem. C 117 (2013) 14588-14599. |
| [29] |
D.A. Svintsitskiy, A.P. Chupakhin, E.M. Slavinskaya, et al., J. Mol. Catal. A:Chem. 368-369 (2013) 95-106. DOI:10.1016/j.molcata.2012.11.015 |
| [30] |
S. Zhu, X. Li, J. Kang, X. Duan, S. Wang, Environ. Sci. Technol. (2018) 307-315. |
| [31] |
Y. Feng, W. Qing, L. Kong, et al., Water Res. 149 (2018) 1-8. |
| [32] |
C. Chen, Y. Zheng, Y. Zhan, et al., Cryst. Growth Des. 8 (2008) 3549-3554. DOI:10.1021/cg7011843 |
| [33] |
S. Sun, C. Li, D. Zhang, Y. Wang, Appl. Surf. Sci. 333 (2015) 229-234. DOI:10.1016/j.apsusc.2015.02.018 |
| [34] |
D. Gao, G. Yang, J. Li, et al., J. Phys. Chem. C 114 (2010) 18347-18351. DOI:10.1021/jp106015t |