 2020, Vol. 31
2020, Vol. 31 


b College of Chemistry, Chemical and Environmental Engineering, Henan University of Technology, Zhengzhou 450001, China;
c State Key Laboratory of Environment-Friendly Energy Material, Southwest University of Science and Technology, Mianyang 621010, China
Metalloenzyme, such as hydrogenases (H2ases), acetyl coenzyme A synthase (ACS), nitrogenases, speed up a variety of reactions that are attractive to theoretical study and industrial application [1]. The mimic of the active sites in various metalloenzyme is an attractive topic in the field of science (Scheme 1). The chemists can be inspired by the natural metabolic processes, and the biologists can learn from the research of the same reactions catalyzed by synthetic model complexes.

|
Download:
|
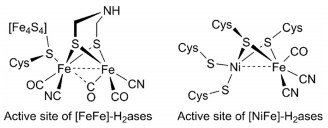
| Scheme 1. Active site of [FeFe]-H2ases and [NiFe]-H2ases. | |
{kind=link}
In the reported works about mimicking the active site of [FeFe]-H2ases, most of model complexes were synthesized by introducing functional ligands into the classical diiron dithiolate complexes[(μ-SR)2Fe2(CO)6] [2]. However, the mimic of [NiFe]-H2ases is more challenging due to the different coordination environments for the two metal centers [3]. Firstly, the structural analogues of these two distinct metal units were synthesized, respectively. Then, the two model complexes were bridged through suitable linkages. Due to the close resemblance with Ni(Scystein)4 site of [NiFe]-H2ases, a various of NiN2S2 and NiS4 metallothiolate complexes were choose as S-donor ligands to bind with the other mononuclear organometallic complexes, such as M(CO)n (M = Mn, Ru), Cp*M (M = Fe, Co, Ru, Rh, Ir), (C6Me6)Ru, Fe[P(OR)3]3 [4]. The synthesized model complexes not only possess the structure characters of active sites in [NiFe]-H2ases, but also reappear their fascinating catalytic abilities [5].
In this work, we reported the synthesis of an air-stable dinickel complex [Ni(phma)(μ-S, S')Ni(dppp)] (1) bearing a NiN2S2 metallothiolate ligand. The thiolate group link two nickel atoms as a bridge, and this structure mimic the principal feature of the active site of [NiFe]-H2ases. In addition, the redox property and particularly electrocatalytic H2-evoluting activity of 1 were investigated.
Reaction of [NEt4]2[Ni(phma)] with one equivalent of [NiCl2(dppp)] in acetonitrile at room temperature gave the desired dinuclear complex [Ni(phma)(μ-S, S')Ni(dppp)] (1) in good yield. The crystal structure of 1 (Fig. 1) shows that both nickel centers display a slightly distorted square planar geometry. The two coordination planes are folded along the μS-μS vector. The dihedral angle defined by the two Ni(μ-S)2 planes is 103.57°. Ni(2) is ligated by the two phosphorus atoms from the dppp ligand and the two sulfur atoms of the phma ligand. And Ni(2) lies 0.012 Å from the P2S2 plane. The Ni(2)PC3P cyclohexane ring is in the chair conformation. Ni(1) ligated by the two nitrogen and two sulfur atoms of the phma ligand. The NiN2S2 unit is essentially planar with a 5-5-5 ring pattern, while Ni(1) lies 0.134 Å from the plane. The Ni(1)–S (2.13 Å) are shorter Ni(2)–S (2.28 Å). The terminal N–Ni–N angle is constrained in the present structure to the square 86.65(13)°. The Ni…Ni distance is 2.6793(7) Å. It has been reported that such a distance is indicative of a Ni–Ni interaction [6].

|
Download:
|
| Fig. 1. Synthesis and molecular structure diagram of complex 1. (All hydrogen atoms and one CH2Cl2 molecular are omitted for clarity). Selected bond length (Å), bond angles and plane angles (°): Ni(1)–N(1) 1.846(3); Ni(1)–N(2) 1.847(3); Ni(1)–S(1) 2.1241(11); Ni(1)–S(2) 2.1327(10); Ni(2)–S(1) 2.2974(11); Ni(2)–S(2) 2.2700(10); Ni(2)–P(1) 2.2003(10); Ni(2)–P(2) 2.1806(11); N(1)–Ni(1)–N(2) 86.65(13); N(1)–Ni(1)–S(1) 92.34(10); N(2)–Ni(1)–S(2) 92.68(10); S(1)–Ni(1)–S(2) 87.36(4); P(1)–Ni(2)–S(1) 93.67(4); P(2)–Ni(2)–S(2) 96.92(4); P(2)–Ni(2)–P(1) 89.40(4); Ni(1)–S(1)–Ni(2) 74.48(4); Ni(1)–S(2)–Ni(2) 74.90(3). | |
{kind=link}
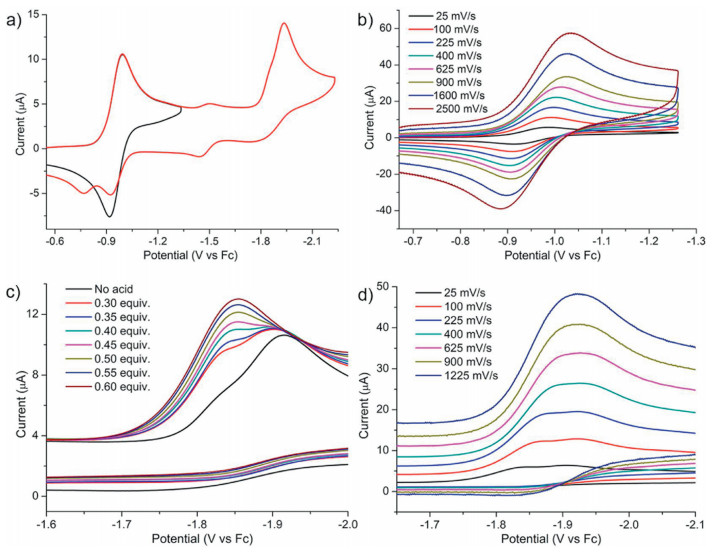
The reduction of 1 was firstly investigated with cyclic voltammetry in DMF. The cyclic voltammogram (CV) exhibits one redox couple at -0.95 V vs. ferrocene (Fc) followed by an irreversible peak at -1.93 V (Fig. 2a). When the cathodic scan is limited to -1.35 V to avoid the second reduction event, the reversibility of thefirst couple was obviously enhanced [7]. The peak-to-peak separation (ΔEp) of the first redox couple is 72 mV (ipa/ipc ≈ 1) at a scan rate of 100 mV/s, indicating a reversible one-electron transfer process [8]. This redox process was ascribed tothe one-electron reduction of NiII/NiI couple in NiP2S2 site [9]. Whatever the scan rate used in the 25–2500 mV/s range, this process indeed appears always chemically reversible (Fig. 2b and Fig. S2 in Supporting information). The variable-scan rate studies prove that this NiII/NiI reduction event is a diffusioncontrolled process. The irreversible process at -1.93 V cannot be explicitly assigned to the reduction of NiI/Ni0 at NiP2S2 moiety or NiII/NiI at NiN2S2 moiety. Hegg reported that the redox of NiII/NiI couple for Ni[N, N'-ethylenebis(2-methylmercaptoacetamide)] located at -1.95 V as a quasireversible wave [10]. More likely, the irreversible peak would be ascribed to the reduction of NiII/NiI at NiN2S2 moiety.

|
Download:
|
| Fig. 2. (a) CVs of 1 (1 mmol/L) recorded in a DMF solution with 0.1 mol/L Bu4NPF6 as the electrolyte at scan rate of 100 mV/s. (b) First reversible redox couple of 1 (1 mmol/L) in 0.1 mol/L [NBu4][PF6]/DMF at various scan rates. (c) CVs of 1 (1 mmol/L) recorded in DMF (0.1 mol/L Bu4NPF6) with the addition of various amounts of ClCH2COOH (0–0.6 mmol/L) at scan rate of 100 mV/s. (d) CVs of 1 (1 mmol/L) recorded in DMF (0.1 mol/L Bu4NPF6) with 0.4 equiv. ClCH2COOH at scan various rates (25–1225 mV/s). | |
{kind=link}
The electrocatalytic performance of 1 for the H2 evolution was evaluated in DMF using ClCH2COOH (pKaDMF = 10.0; E°HA = -1.36 V vs. Fc) as proton source [11]. To emphasize an important point, complex 1 is quite stable in the presence of excess ClCH2COOH, as monitored by UV–vis spectrometry (Fig. S9 in Supporting information). Upon the addition of 0.3 equiv. acid, a new reduction event appears at about -1.86 V (Fig. 2c), which is ascribed to a prewave of total catalysis (zone KT2, pure kinetic conditions and total catalysis) [12]. In this case, complex 1 consumes all acids local to the electrode surface resulting in a CV with two peaks. An irreversible catalytic peak is initially observed at positive potential of 1-/12-. As the potential is scanned more negative, the reduction of 1-/12- redox is still seen in the same position with no acid present. Subsequently, the addition of 0.6 equiv. ClCH2COOH leads an overlap of these two peaks indicating the transition from KT2 into K zone [12]. Between 0.7 equiv. and 6 equiv. of ClCH2COOH, the catalytic current (icat1) slowly grows with increasing of acid (Fig. S3 in Suppporting information). We propose this is an inefficient H2 evolution reaction. The peak potential and peak current of total catalysis waveforms notonly depend on the substrate concentration, but also relate to the scan rate. Increase of scan rate could also push the CV response into zone K. In the present of 0.4 equiv. ClCH2COOH, when the scan rate is above 900 mV/s, the prewave shifts to more negative potentials erasing the distinction of these two peaks (Fig. 2d).
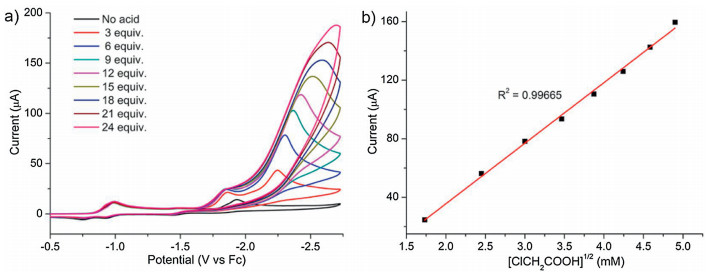
When the potential is scanned more negative, a remarkable enhancement of catalytic current (icat2) are observed at about -2.25 V indicating another H2-evoluting process (Fig. 3a). The standard rinse experiments excluded that the electrocatalysis is initiated by the electrodeposits of an ill-defined nanoparticulate material at the electrode surface (Fig. S7 in Suppporting information). The linear correlation between icat2 and the square root of acid concentration indicates that the observed rate constant for catalysis is first-order in acid (Fig. 3b). Furthermore, icat2 also shows varied linearly with the square root of scan rate (Fig. S6 in Suppporting information), implying that the electrocatalytic process is diffusion controlled. The overpotential (η) of 0.800.97 V for electrocatalytic proton reduction is given by calculating the difference between the catalytic half-wave potential (Ecat/2) and the standard potential for the formation of H2 from ClCH2COOH (E°HA) [13]. On the basis of the electrochemical data, the mechanism of the electrocatalytic H2 evolution is proposed to be an ECCE process starting from 1- [14]. But with side phenomena, such as consumption of the substrate, deactivation of the catalyst, inhibition of the current by H2 adsorption to the electrode, and background H2 evolution, no matter how to increase the scan rate or acid concentration, the catalytic response currents of complex 1 does not show idealized S-type with fixed plateau current [15]. Therefore, we cannot get convincing kinetic information for this electrocatalytic H2-evoluting process via the ratio of icat/ip and the foot-of-the-wave method [16]. As emphasized by Dempsey et al., it should be careful to use the ratio of icat/ip to calculate the rate constant (kobs) of electrocatalytic H2-evoluting reaction [13]. The applications of eqs. 2 and 5 are restricted to the S-shaped catalytic waves of zones KS and KD. Before the calculation, it also needs to ensure that an appropriate scan rate is being utilized. With the precondition Eq. 2, icat should be independence on the scan rate [17].

|
Download:
|
| Fig. 3. CVs of 1 (1 mmol/L) recorded in DMF (0.1 mol/L Bu4NPF6) with the addition of various amounts of ClCH2COOH (0–24 mmol/L) at scan rate of 100 mV/s. (b) Plot of icat/ip vs. [ClCH2COOH]1/2. | |
{kind=link}
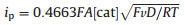
|
(1) |
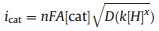
|
(2) |
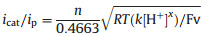
|
(3) |

|
(4) |
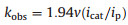
|
(5) |
Controlled potential electrolysis (CPE) experiments were performed at -1.83 V and -2.43 V (vs. Fc), respectively, which provide further information for the electrocatalytic H2 evolution with 1. After electrolysis over a 3 h period at -2.43 V, 7.44 μmol H2 was detected by gas chromatography, thus giving a Faradaic efficiency of 72% and a turnover number (TON) of 1.2. UV–vis spectroscopy showed that 65% of 1 was still in the solution. (Fig. S10 in Supporting information) Electrolyzing the identical solution at -1.83 V resulted in the formation of 1.80 μmol H2, giving a reduced TON of only 0.3.
In summary, the metallothiolate [Ni(phma)]2- was used as cisdithiolate sulfur donor to react with [NiCl2(dppp)], affording an airstable dinickel complex 1, which not only possess the principal feature of the active site of [NiFe]-H2ases, but also reappear their fascinating catalytic abilities. When the redox property was investigated via CV and CPE experiments, 1 was verified to be a moderate electrocatalysts of H2-evoluting reaction using ClCH2COOH as the proton source.
AcknowledgmentsThis work was supported by National Natural Science Foundation of China (Nos. 21773184, 21671158 and 21601164), Natural Science Foundation of Henan (No.162300410052), Key Science and Technology Project of Henan (No. 172102310137).
Appendix A. Supplementary dataCCDC 1942436 and 1942438 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336, 033. Supplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.01.033.
| [1] |
(a) M. Can, F.A. Armstrong, S.W. Ragsdale, Chem. Rev. 114(2014) 4149-4174; (b) T.R. Simmons, G. Berggren, M. Bacchi, M. Fontecave, V. Artero, Coord. Chem. Rev. 270-271(2014) 127-150; (c) Y.L. Hu, A.W. Fay, C.C. Lee, J. Yoshizawa, M.W. Ribbe, Biochemistry 47(2008) 3973-3981. |
| [2] |
(a) Y.L. Li, T.B. Rauchfuss, Chem. Rev. 116(2016) 7043-7077; (b) J.F. Capon, F. Gloaguen, F.Y. Pétillon, P. Schollhammer, J. Talarmin, Coord. Chem. Rev. 253(2009) 1476-1494; (c) I.P. Georgakaki, L.M. Thomson, E.J. Lyon, M.B. Hall, M.Y. Darensbourg, Coord. Chem. Rev. 238-239(2003) 255-266; (d) N. Wang, M. Wang, L. Chen, L. Sun, Dalton Trans. 42(2013) 12059-12071; (e) Y. Zhao, X. Yu, H. Hu, et al., Chin. Chem. Lett. 29(2018) 1651-1655. |
| [3] |
(a) S. Ogo, Coord. Chem. Rev. 334(2017) 43-53; (b) D.J. Evans, C.J. Pickett, Chem. Soc. Rev. 32(2003) 268-275; (c) J.A. Denny, M.Y. Darensbourg, Chem. Rev. 115(2015) 5248-5273; (d) D. Schilter, J.M. Camara, M.T. Huynh, S. Hammes-Schiffer, T.B. Rauchfuss, Chem. Rev. 116(2016) 8693-8749. |
| [4] |
(a) S. Ogo, R. Kabe, K. Uehara, et al., Science 316(2007) 585-587; (b) S. Ogo, K. Ichikawa, T. Kishima, et al., Science 339(2013) 682-684; (c) D. Brazzolotto, M. Gennari, N. Queyriaux, et al., Nat. Chem. 8(2016) 1054-1060; (d) W. Zhu, A.C. Marr, Q. Wang, et al., PNAS 102(2005) 18280-18285; (e)V.Fourmond, S.Canaguier, B.Golly, etal., EnergyEnviron.Sci.4(2011)2417-2427; (f) S. Ding, P. Ghosh, A.M. Lunsford, et al., J. Am. Chem. Soc. 138(2016) 12920-12927; (g) D.H. Manz, P.C. Duan, S. Dechert, et al., J. Am. Chem. Soc. 139(2017) 16720-16731. |
| [5] |
(a) D. Brazzolotto, L. Wang, H. Tang, et al., ACS Catal. 8(2018) 10658-10667; (b) Y.X.C. Goh, H.M. Tang, W.L.J. Loke, W. Fan, Inorg. Chem. 58(2019) 12178-12183; (c) C.U. Perotto, C.L. Sodipo, G.J. Jones, et al., Inorg. Chem. 57(2018) 2558-2569; (d) G. Gezer, S. Verbeek, M.A. Sieglerb, E. Bouwman, Dalton Trans. 46(2017) 13590-13596; (e) B.E. Barton, C.M. Whaley, T.B. Rauchfuss, D.L. Gray, J. Am. Chem. Soc. 131(2009) 6942-6943; (f) L. Song, Y. Lu, L. Zhu, Q.L. Li, Organometallics 36(2017) 750-760; (g) P. Sun, D. Yang, Y. Li, et al., Organometallics 35(2016) 751-757; (h) O.A. Ulloa, M.T. Huynh, C.P. Richers, et al., J. Am. Chem. Soc. 138(2016) 9234-9245; (i) K. Weber, T. Krämer, H.S. Shafaat, et al., J. Am. Chem. Soc. 134(2012) 20745-20755; (j) X. Chu, J. Jin, B. Ming, et al., Chem. Sci. 10(2019) 761-767. |
| [6] |
(a) M.A. Turner, W.L. Driessen, J. Reedijk, Inorg. Chem. 29(1990) 3331-3335; (b) P.V. Rao, S. Bhaduri, J. Jiang, R.H. Holm, Inorg. Chem. 43(2004) 5833-5849. |
| [7] |
(a) N. Wang, M. Wang, Y. Wang, et al., J. Am. Chem. Soc. 135(2013) 13688-13691; (b) D. Zheng, N. Wang, M. Wang, et al., J. Am. Chem. Soc. 136(2014) 16817-16823; (c) D. Li, C.N. Lin, S.Z. Zhan, C.L. Ni, Chin. Chem. Lett. 28(2017) 1424-1428. |
| [8] |
A.J. Bard, L.R. Faulkner, Electrochemical Methods:Fundamentals and Applications, 2nd ed. 2nd ed. New York: Wiley, 2001.
|
| [9] |
(a) S.E. Duff, J.E. Barclay, S.C. Davies, D.J. Evans, Inorg. Chem. Commun. 8(2005) 170-173; (b) D.J. Evans, Eur. J. Inorg. Chem. 22(2005) 4527-4532. |
| [10] |
Ø. Hatlevik, M.C. Blanksma, V. Mathrubootham, A.M. Arif, E.L. Hegg, J. Biol. Inorg. Chem. 9 (2004) 238-246. DOI:10.1007/s00775-003-0518-8 |
| [11] |
G.A.N. Felton, R.S. Glass, D.L. Lichtenberger, D.H. Evans, Inorg. Chem. 45 (2006) 9181-9184. DOI:10.1021/ic060984e |
| [12] |
(a) J.M. Savéant, K.B. Su, J. Electroanal. Chem. 191(1984) 341-349; (b) E.S. Rountree, B.D. McCarthy, T.T. Eisenhart, J.L. Dempsey, Inorg. Chem. 53(2014) 9983-10002; (c) K.J. Lee, B.D. McCarthy, E.S. Rountree, J.L. Dempsey, Inorg. Chem. 56(2017) 1988-1998. |
| [13] |
A.M. Appel, M.L. Helm, ACS Catal. 4 (2014) 630-633. DOI:10.1021/cs401013v |
| [14] |
C. Costentin, J.M. Savéant, Chem. Electro. Chem. 1 (2014) 1226-1236. |
| [15] |
C. Costentin, S. Drouet, M. Robert, J.M. Savéant, J. Am. Chem. Soc. 134 (2012) 11235-11242. DOI:10.1021/ja303560c |
| [16] |
C.P. Yap, K. Hou, A.A. Bengali, W.Y. Fan, Inorg. Chem. 56 (2017) 10926-10931. DOI:10.1021/acs.inorgchem.7b01079 |
| [17] |
(a) M.L. Helm, M.P. Stewart, R.M. Bullock, M.R. DuBois, D.L. DuBois, Science 333(2011) 863-866; (b) R. Tatematsu, T. Inomata, T. Ozawa, H. Masuda, Angew. Chem. Int. Ed. 55(2016) 5247-5250. |