 2020, Vol. 31
2020, Vol. 31 


The daphniphyllum alkaloids are a unique group of azapolycyclic natural products (> 320 members) that are found in the genus daphniphyllum. Based on their different polycyclic skeletons, daphniphyllum alkaloids were classified into 20 types [1]. Calyciphylline A-type [2], daphnicyclidin-type [3] and macropodumine-type [4] daphniphyllum alkaloids were biosynthesized from a common precursor proto-daphniphylline and possess a similar 5/5 AE bicyclic system [5]. The AE bicyclic can be considered as the core structure of these alkaloids because it fused with all of the rest rings and contain most of the chiral centers and quaternary carbons. The representative examples of those alkaloids are depicted in Fig. 1. Much attention has been attracted to the synthesis of them due to their complex polycyclic skeletons and wide range of biological activities, including antitumor, antiviral, and nerve growth factor-regulating properties [6]. To date, the total synthesis of several calyciphylline A-type alkaloids have been elegant accomplished by developing highly efficient synthesis strategies [7]. However, due to the construction of seven-membered ring C of daphnicyclidin-type is more a challenge than the six-membered ring C in calyciphylline A-type, the total synthesis of daphnicyclidin-type was not reported so far, only a few reports on the synthesis of its partial ring system [8]. Herein, we disclose an efficient synthesis of the ACE tricyclic systems of daphnicyclidin A and dehydroxymacropodumine A via a decarboxylation radical conjugate addition reaction [9].

|
Download:
|
| Fig. 1. Representative structures of calyciphylline A-type, daphnicyc-lidin-type and macropodumine-type alkaloids | |
{kind=link}
In our previous work, we have developed an efficient approach to access fast and stereoselective construction of 2, 3, 4-cis trisubstituted pyrrolidine 1 via tandem N-allylation/SN2' reaction [8d]. Methylation of the α-position of lactone of 1, followed by hydrolyzing the lactone, a carboxyl group at the 3-position could be delivered, which will produce a tertiary carbon radical under the catalysis of iridium or ruthenium complex. Consequently, it is reasonable that if the new generated free radical was captured by a cyclopentenone, the AE bicyclic system will be constructed.
Our retrosynthesis is described in Scheme 1, ring D of daphnicyclidin A could be constructed by a SmI2-mediated radical Michael addition [10] from intermediate a, and the 11-membered macrolide ring of dehydroxymacropodumine A could be assembled by macrolactonization [11] from intermediate b. The ring B of those two alkaloids can be formed by the formation of lactam. Finally, Seven-membered ring C and C-5 quaternary carbon center can be constructed via a photocatalyzed decarboxylation radical conjugate addition from 20 and 27 respectively, which can be synthesized from 2, 3, 4-cis tri-substituted pyrrolidine 1.

|
Download:
|
| Scheme 1. Retrosynthesis of daphnicyclidin A and dihydroxy-macropodumine A | |
{kind=link}
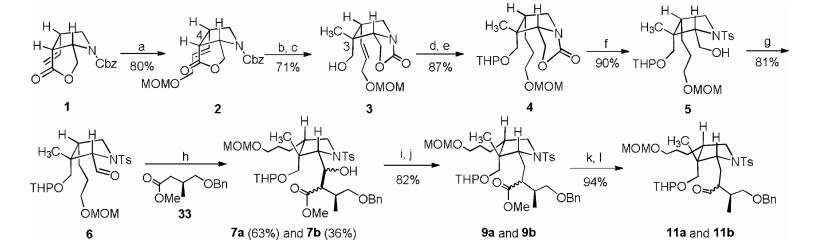
We first modified the substituents at the 2, 3, 4-position of 1. As showed in Scheme 2, under the catalysis of Grubbs(II) catalyst, the 4-vinyl group of 1 react with MOM protected allyl alcohol give 2 in 80% yield. Treatment of 2 with LiHMDS and MeI in THF at -78 to 45 ℃, the product of C-3 methylation was delivered with no formation of undesired isomer. Hydrolyzing the lactone of 2a with sodium hydroxide in MeOH/H2O results in the formation of oxazolinone carboxylic acid sodium salt. After removing MeOH/H2O, the residue was added THF followed by ethyl chloroformate, a mixed-anhydride was produced which was reduced by NaBH4 to give alcohol 3 in 71% yield. Protecting the alcohol with THP and reduce the double bond with 10% Pd/C under 1 atm of hydrogen afford 4 in 87% yield. The following hydrolysis of the oxazolinone with t-BuOK in aqueous t-BuOH at 100 ℃ deliver a 2-hydroxymethyl pyrrolidine intermediate, which react with p-TsCl by adjusting pH of the reaction solution to 8–9 give 5 in 90% yield. Subsequently oxidation of the primary alcohol with Dess-Martin reagent produces aldehyde 6 for further aldol reaction. Treatment of ester 33 with LiHMDS in THF at 78 ℃ followed by addition of aldehyde 6 smoothly deliver secondary alcohol 7a and 7b in 63% and 36% yield, respectively. The hydroxyl of 7a and 7b was removed by Barton deoxygenation [12] give 9a and 9b in 82% yield. Convention of the ester group of 9a and 9b into alcohol with LiAlH4 in THF at 20 ℃ then oxidize the resulting alcohol with NMO/TPAP give aldehyde 11a and 11b as a pair of diastereomers.

|
Download:
|
| Scheme 2. Synthesis of aldehyde 11a and 11b.Reagents and conditions: (a) MOMOCH2CH = CH2, Grubbs(II) (3.0%), CH2ClCH2Cl, 70 ℃, 48 h; (b) LiHMDS, CH3I, THF, 78 ℃ to 45 ℃; (c) NaOH (2.0 eq.), MeOH:H2O = 4:1, 25 ℃, then THF, 0 ℃, ClCOOEt, then NaBH4; (d) PPTs, DHP, DCM, 25 ℃, 8 h; (e) Pd/C (10.0%), H2, 1 atm, MeOH, 25 ℃, 2 h; (f) t-BuOK, t-BuOH:H2O = 10:1, 100 ℃, then HCl (1 mol/L) (pH 8–9), then TsCl, 25 ℃, 5 h; (g) NMO, TPAP, DCM, 35 ℃, 10 min; (h) LiHMDS, THF, 78 ℃, 33; (i) CS2, NaH, THF, 0 ℃–25 ℃, 8 h, then MeI, 25 ℃, 2 h; (j) n-Bu3SnH, AIBN, PhMe, 105 ℃, 5 h; (k) LiAlH4, THF, -20 ℃, 2 h; (l) NMO, TPAP, DCM, 35 ℃, 10 min. | |
{kind=link}
The determination of the stereochemistry of 11a and 11b was conducted by conversion of 11a and 11b into 6-hexanolactone 13a and 13b by deprotecting the THP group then oxidation of the resulting hemiacetal to lactone. The 1H and 13C NMR signal of 13a and 13b were assigned by detailed analyses of 1D and 2D NMR spectra (Tables S1 and S2 in Supporting information). In the NOESY spectra of 13a, cross-peak of H-2 (δH 2.46) to H-4 (δH 2.77) and H-2 to H-21 (δH 0.94) was not observed, indicated that the relative configuration of 13a is 2, 4-trans-4, 5-cis (Fig. S1 in Supporting information). Likewise, the 1H and 13C NMR signal of 13b were assigned, the cross-peak H-2 (δH 2.42) to H-4 (δH 3.53) and H-21 (δH 0.75) were observed in NOESY revealed that the relative configuration of 13b is 2, 4, 5-cis (Fig. S2 in Supporting information), which is consistent with that of daphnicyclidin A and dihydroxymacropodumine A. Subsequently, we attempted to change the configuration of the α-position of aldehyde 11a under basic conditions. Treatment of 11a with DBU in CH3CN at 75 ℃, a mixture of 11a and 11b was obtained in a 1:1 ratio. Aldehyde 11a and 11b cannot be separated by column chromatography. Fortunately, alcohols 10a and 10b obtained by reduction of the aldehyde group can be separated by column chromatography. Consequently, oxidation of 10b could give 11b, while 10a can be oxidized to 11a, and then repeated the configuration inversion process (Scheme 3).

|
Download:
|
| Scheme 3. Determination of the stereochemistry of 11a, 11b and transformation of 11a to 10b. Reagents and conditions: (a) p-TSA, acetone:H2O = 4:1, 25 ℃, 24 h; (b) Jones' reagent, acetone, 0℃, 10 min; (c) DBU, CHCN, 75 ℃, 12 h, then NaBH4, H2O, 25 ℃, 15 min; (d) NMO/TPAP, DCM. | |
{kind=link}
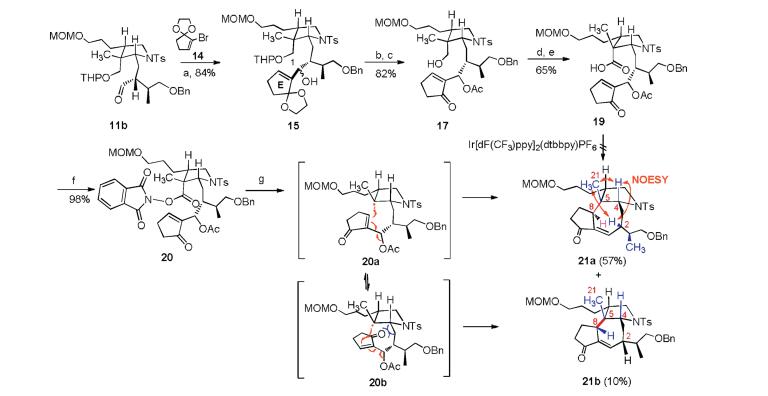
With 11b in hand, we first attempted to attach the cyclopentenone to the aldehyde 11b by MBH reaction [13], but it was unsuccessful. Therefore, we converted cyclopentenone to alkenyl bromide 14, as showed in Scheme 4, treatment of 14 with t-BuLi in THF at 78 ℃, followed by addition of 11b, the product 15 was delivered in 85% yield. Protection of the new formed secondary alcohol with acetyl group and removed the ethylene glycol and THP protecting group to give a primary alcohol 17 in 82% yield. Conversion of the hydroxyl group to aldehyde with NMO/TPAP then oxidation of the resulting aldehyde with NaH2PO4/NaClO2 give the carboxylic acid 19, which was subjected to the decarboxylation radical conjugate addition using MacMillan's condition [9a]. However, this reaction is complicated, and the conjugate addition products were not isolated. We next converted the carboxyl group to N-hydroxyphthalimide ester to give 20 and try Overman's condition [9b, 9c]. Using [Ru(bpy)3](BF4)2 as catalyst, diethyl 1, 4-dihydro-2, 6-dimethyl-3, 5-pyridinedicarboxylate as photoelectron transfer agent, DIPEA as a base and deoxygenated DCM as solvent, under the irradiation of blue light, the product 21a and 21b was obtained in a 67% yield. Compounds 21a and 21b are a pair of inseparable diastereoisomers in a 5.7:1 ratio. In the 1H NMR of 21a and 21b, the proton signal of double bond at d = 6.54 (major) and 6.57 (minor) ppm were observed and the proton signal of acetyl group were absent, indicated that the acetyl group was kicked off after the conjugate addition. The molecular formula of 21a and 21b was established as C35H47NO6S by HRMS at m/z 632.3011 [M+Na]+ (calcd. 632.3016), further confirming the absence of acetoxyl group. The 1H and 13C NMR signal of the major product 21a were assigned by detailed analyses of 1D and 2D NMR spectra (Table S3 in Supporting information). In the NOESY spectra of 21a, the cross-peak of H-8 (δH 3.36) to H-2 (δH 3.27), H-4 (δH 3.49) or H-21 (δH 0.86) were not observed, indicated that the relative configuration of 21a is 2, 4, 5-cis-5, 8-trans (Fig. S3 in Supporting information). It is believed that the diastereoselectivity shown in this reaction is due to the steric hindrance of transition state 20a is less than 20b. The 1H and 13C signals of 20b are fail to be assigned because most of its NMR signals are overlap with 21a.

|
Download:
|
| Scheme 4. Construction of ACE tricyclic of daphnicyclidin A by decarboxylation conjugate addition. Reagents and conditions: (a) 14, t-BuLi, THF, 78 ℃; (b) Ac2O, Et3N, DMAP, DCM, 25 ℃, 2 h; (c) p-TSA, acetone:H2O = 4:1, 35 ℃, 12 h; (d) NMO, TPAP, DCM, 35 ℃; (e) NaH2PO4, NaClO2, t-BuOH/CH3CN/H2O = 2:2:1, 0 ℃–25 ℃; (f) N-hydroxyphthalimide, DCC, THF, 25 ℃, 24 h; (g) Diethyl 1, 4-dihydro-2, 6-dimethyl-3, 5-pyridinedicarboxylate, CH2Cl2, DIPEA, blue light, [Ru(bpy)3](BF4)2, 25 ℃, 2 h | |
{kind=link}
After successfully construction of the ACE tricyclic system of daphnicyclidin A, using the same strategy, we studied the synthesis of the ACE tricyclic system of dehydroxymacropodumine A. As showed in Scheme 5, treatment of 22 with tBuLi in THF at 78 ℃ followed by addition of 11b, a cyclopentene was attached to afford secondary alcohol 23 in 81% yield. Oxidation of the alcohol with Dess-Martin reagent, deprotection of the THP group and oxidation of the resulting primary alcohol give aldehyde 26 in 73% yield. Further oxidation of the aldehyde group to carboxylic acid gave 27, which was subjected to MacMillan's photocatalyst condition. To our delight, using 1 mol% Ir[dF(CF3) ppy]2(dtbbpy)PF6 as catalyst in deoxygenated DMF, a household 26 W fluorescent light bulb irradiation about 16 h, the conjugate addition product 29 was delivered in 56% yield. We also examined Overman's condition by conversion of carboxylic acid 27 to N-hydroxyphthalimide ester 28. Using the same reaction condition with that of 20 to 21a, compound 29 was also delivered in 52% yield. The molecular formula of29 was established as C37H47NO6S by HRMS at m/z 672.3565 [M+H]+ (calcd. 672.3569), consistent with its structure. The NMR spectra of 29 were complicated because it is a mixture of several diastereoisomers. To further identify the structure of 29, the MOM group was removed and the resulting hydroxyl groups were converted to carbonyl group afforded 30. The molecular formula of 30 was established as C33H41NO6S by HRMS at m/z 602.2554 [M[+Na]+ (calcd. 602.2547). The NMR of 30 indicated that it is mainly a single compound and its 1H and 13C NMR signal were assigned by detailed analyses of 1D and 2D NMR spectra (Table S4 in Supporting inforamtion). The NOESY cross-peaks of H-2 (δH 3.68) to H-4 (δH 3.46), H-4 to H-21 (δH 0.67) and H-2 to H-13 (δH 3.29) showed that H-2, H-4, H-13 were at the convex side and the H-8 (δH 3.18) on the concave side (Fig. S4 in Supporting information). The configuration of C-8 is identical to the configuration of C-8 in compound 21a.

|
Download:
|
| Scheme 5. Construction of ACE tricyclic of dehydroxymacro-podumine A by decarboxylation conjugate addition. Reagents and conditions: (a) 22, t-BuLi, THF, 78 ℃; (b) DMP, DCM, 25 ℃; (c) 1 mol/L HCl, MeOH, 25 ℃, 24 h; (d) DMP, DCM, 25 ℃; (e) NaH2PO4, NaClO2, t-BuOH/CH3CN/H2O = 2:2:1, 0 ℃–25 ℃; (f) N-hydroxy-phthalimide, DIC, THF, 25 ℃, 24 h; (g) 1 mol% Ir[dF(CF3)ppy]2(dtbbpy)PF6, K2HPO4, DMF, 25 ℃, 26 W GFL, 16 h; (h) diethyl 1, 4-dihydro-2, 6-dimethyl-3, 5-pyridine-dicarboxylate, CH2Cl2, DIPEA, blue light, [Ru(bpy)3](BF4)2, 25 ℃, 2 h; (i) 3 mol/L HCl, MeOH, 12 h; (j) DMP, DCM, 25 ℃ | |
{kind=link}
In summary, we have developed a novel synthesis toward the ACE tricyclic cores of daphnicyclidin A and dehydroxy-macropodumine A. The chiral fragment 33 was introduced by aldol reaction for further construction of piperidine ring B and seven membered ring C, the E ring was introduced by nucleophilic addition of lithium pentene to aldehyde and the two ACE tricyclic systems were finally constructed by photocatalytic decarboxylation conjugate addition. The total synthesis of dehydroxymacropodumine A is underway in our laboratory and will be disclosed in due course.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 21372149, 21572125) and the Fundamental Research Funds for the Central Universities of China (No. GK201703023) and Shaanxi Normal University.
Appendix A. Supplementary dataSupplementarymaterial related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.12.018.
| [1] |
(a) A.K. Chattopadhyay, S. Hanessian, Chem. Rev. 117 (2017) 4104-4146; (b) H.F. Wu, X.P. Zhang, L.S. Ding, et al., Planta Med. 79 (2013) 1589-1598; (c) J. Kobayashi, T. Kubota, Nat. Prod. Rep. 26 (2009) 936-962. |
| [2] |
(a) H. Morita, J. Kobayashi, Org. Lett. 5 (2003) 2895-2898; (b) H. Zhang, S.P. Yang, C.Q. Fan, J. Ding, J.M. Yue, J. Nat. Prod. 69 (2006) 553-557; (c) S.P. Yang, H. Zhang, C. Zhang, J.M. Yue, J. Nat. Prod. 69 (2006) 79-82; (d) S.Z. Mu, C.S. Li, H.P. He, J. Nat. Prod. 70 (2007) 1628-1631; (e) Q. Zhang, Y.T. Di, C.S. Li, Org. Lett. 11 (2009) 2357-2359; (f) H. Zhang, S.L. Shyaula, J.Y. Li, J. Nat. Prod. 78 (2015) 2761-2767; (g) H. Zhang, S.L. Shyaula, J.Y. Li, Org. Lett. 18 (2016) 1202-1205. |
| [3] |
(a) H. Morita, N. Yoshida, J. Kobayashi, J. Org. Chem. 67 (2002) 2278-2282; (b) S.P. Yang, J.M. Yue, Org. Lett. 6 (2004) 1401-1404; (c) M.M. Cao, H.P. He, Y.C. Gu, Nat. Prod. Bioprospect. 3 (2013) 29-32; (d) Y. Lu, K. Gao, X. Wang, Molecules 19 (2014) 3055-3067. |
| [4] |
(a) Z.Y. Li, P. Chen, H.G. Xu, et al., Chin. J. Chem. 26 (2008) 348-352; (b) W. Zhang, Y.W. Guo, K. Krohn, Chem.-Eur. J. 12 (2006) 5122-5127. |
| [5] |
(a) C.H. Heathcock, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 14323-14327; (b) Y. Inaba, H. Morita, J. Kobayashi, J. Am. Chem. Soc.123 (2001) 11402-11408; (c) H. Morita, J. Kobayashi, Tetrahedron 58 (2002) 6637-6641; (d) Y.T. Di, X.J. Hao, J. Nat. Prod. 82 (2019) 427-430. |
| [6] |
(a) H. Zhang, S.P. Yang, C.Q. Fan, J. Ding, J.M. Yue, J. Nat. Prod. 69 (2006) 553-557; (b) C.R. Zhang, H.B. Liu, T. Feng, et al., J. Nat. Prod. 72 (2009) 1669-1672; (c) S. Liu, J.H. Zhang, Y.T. Di, J.Y. Dong, X.J. Hao, Nat. Prod. Res. 32 (2018) 2165-2170; (d) J.B. Xu, H. Zhang, L.S. Gan, et al., J. Am. Chem. Soc. 136 (2014) 7631-7633; (e) H. Morita, N. Ishioka, H. Takatsu, et al., Org. Lett. 7 (2005) 459-462; (f) S. Saito, H. Yahata, T. Kubota, et al., Tetrahedron 64 (2008) 1901-1908. |
| [7] |
(a) Z. Lu, Y. Li, J. Deng, A. Li, Nat. Chem. 5 (2013) 679; (b) J. Li, W. Zhang, F. Zhang, A. Li, J. Am. Chem. Soc. 139 (2017) 14893-14896; (c) Y. Chen, W. Zhang, L. Ren, A. Li, Angew. Chem. Int. Ed. 57 (2018) 952-956; (d) H. Shi, I.N. Michaelides, B. Darses, D. Darren, J. Am. Chem. Soc. 139 (2017)17755-17758; (e) X. Chen, H.J. Zhang, X. Yang, H.B. Zhai, Angew. Chem. Int. Ed. 130 (2018)959-963; (f) W. Zhang, M. Ding, J. Li, A. Li, J. Am. Chem. Soc. 140 (2018) 4227-4231; (g) H.Y. Sun, G.M. Wu, X.G. Xi, Chin. Chem. Lett. 30 (2019) 1538-1540; (h) H.J. Wang, Q.Y. Dong, Q.X. Xie, P. Tang, Chin. Chem. Lett. 31 (2019) 685-688. |
| [8] |
(a) S. Ikeda, M. Shibuya, N. Kanoh, Y. Iwabuchi, Org. Lett. 11 (2009) 1833-1836; (b) T.B. Dunn, J.M. Ellis, C.C. Kofink, L.E. Overman, Org. Lett. 11 (2009) 5658-5661; (c) D.R. Williams, P.K. Mondal, S.A. Bawel, Org. Lett. 16 (2014) 1956-1959; (d) J.L. Li, H.W. Shi, Q. Wang, J. Yang, Org. Lett. 19 (2017) 1497-1499. |
| [9] |
(a) O. Chisa, W.C. David, MacMillan, J. Am. Chem. Soc.136 (2014) 10886-10889; (b) M.J. Schnermann, L.E. Overman, Angew. Chem. Int. Ed. 51 (2012) 9576-9580; (c) J.T. Daniel, S. Yuriy, M. Mikko, L.E. Overman, J. Am. Chem. Soc. 140 (2018)3091-3102; (d) Y. Ouyang, Y. Peng, W.D.Z. Li, Tetrahedron 75 (2019) 4486-4496. |
| [10] |
(a) A. Nakazaki, T. Era, Y. Numada, Tetrahedron 62 (2006) 6264-6271; (b) H. Yu, T. Gai, W.L. Sun, M.S. Zhang, Chin. Chem. Lett. 22 (2011) 379-381; (c) M. Gao, Y.C. Wang, Z.H. Yao, Angew. Chem. Int. Ed. 57 (2018) 13313-13318. |
| [11] |
(a) E.J. Corey, K.C. Nicolaou, J. Am. Chem. Soc. 96 (1974) 5614-5616; (b) M.B. Andrus, T.L. Shih, J. Org. Chem. 61 (1996) 8780-8785; (c) X.M. Zhu, L.L. He, G.L. Yang, et al., Synlett. (2006) 3510-3512;(d) R.G. Ren, J.Y. Ma, Z.Y. Mao, Y.W. Liu, B.G. Wei, Chin. Chem. Lett. 26 (2015)1209-1215. |
| [12] |
(a) D.H.R. Barton, S.W. McCombie, J. Chem. Soc. 1 (1975) 1574-1585; (b) H. Deng, X. Yang, Z. Tong, Z. Li, H. Zhai, Org. Lett. 10 (2008) 1791-1793; (c) Z.H. Zhao, A. Wang, P.S. Lei, Chin. Chem. Lett. 30 (2019) 425-427. |
| [13] |
(a) Y.H. Wan, A. Shaik, K. Chen, Chem. Rec. 17 (2017) 363-381; (b) D. Daniel, R. Oliver, J. Org. Chem. 81 (2016) 10357-10365; (c) H.Y. Duan, J. Ma, Z.Z. Yuan, F. Xu, Chin. Chem. Lett. 26 (2015) 646-648. |