 2020, Vol. 31
2020, Vol. 31 


C19-diterpenoid alkaloids are a large class of pseudo-alkaloids that constitute the main chemical components in plants of the genus Aconitum [1]. Many of these compounds exhibited antiinflammatory, analgesic, and antiarrhythmic effects. A majority of the C19-diterpenoid alkaloids (e.g., 1, Fig. 1) have been categorized as the aconitine-type molecules (named by the representative member aconitine [2]), the structures of which are characterized by a complex and rigid hexacyclic skeleton and densely oxygenated substituents. In 2008, Tan and colleagues isolated a rearrangedtype C19-diterpenoid alkaloid named vilmoraconitine (2, Fig. 1) from Aconitum vilmorinianum [3]. Structurally, this unique compound possesses an additional C8–C10 bond, thus forming a highly congested cyclopropane ring (C8, C9 and C10) compared to the aconitine-type counterparts in the C19-diterpenoid alkaloid family. Later, several natural congeners (e.g., 3–5, Fig. 1) of the rearranged-type alkaloids were found from related Aconitum species [4].

|
Download:
|
| Fig. 1. Structures of the aconitine- and rearranged-type C19-diterpenoid alkaloids. | |
{kind=link}
Due to their intricate architectures, C19-diterpenoid alkaloids have challenged synthetic chemists for decades [5]. Wiesner and co-workers made pioneering contributions in this field [6], who achieved the total synthesis of three aconitine-type alkaloids talatisamine [6a], chasmanine [6b], and 13-desoxydelphonine [6c] in the 1970s. Recently, the groups of Gin [7], Sarpong [8], Fukuyama [9]and Inoue [10] independently reported successful approaches to related norditerpenoid alkaloids, while many other groups developed a number of synthetic strategies for construction of various ring systems [11]. Despite these advances, chemical synthesis of the aforementioned rearranged-type C19-diterpenoid alkaloids (i.e., 2–5, Fig. 1), to the best of our knowledge, has been unknown. Apart from the polycyclic and bridged framework as well as diverse substitution groups, in particular, the unusual cyclopropane unit bearing in this type of alkaloids poses additional synthetic challenges [12]. Intrigued by the fascinating structure of the rearranged-type alkaloids and in combination with the continuing pursuit of various diterpenoid alkaloids in our laboratory [13], we launched a research program toward the total synthesis of vilmoraconitine (2). Herein, we report our efforts that led to an expedient assembly of the 6/3/5/6 tetracyclic core of the target alkaloid 2.
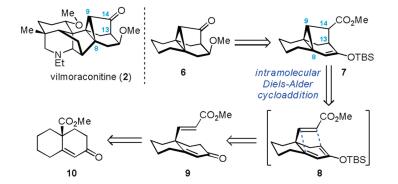
Illustrated in Scheme 1 is the retrosynthesis of the 6/3/5/6 tetracyclic core (6) of vilmoraconitine. Compound 6 could be prepared from the enol silane 7 through simple functional group manipulations. The key cyclopropane ring in 7 was envisioned to be formed by means of an intramolecular Diels–Alder cycloaddition of triene 8 [14]. Notably, such a transformation would allow us to establish the complete 6/3/5/6 tetracyclic core via construction of the C8–C9 and C13–C14 bonds in a concerted way. In turn, triene 8 was accessible from enone 9 and the latter could be synthesized from the known bicyclic ester 10 [15].

|
Download:
|
| Scheme 1. Retrosynthesis of the 6/3/5/6 tetracyclic core (6) of vilmoraconitine. | |
{kind=link}
Execution of the above synthetic plan resulting in the successful access to the tetracyclic fragment 12 is depicted in Scheme 2. The known bicyclic enone 10 was easily prepared in racemic form from the commercially available cyclohexanone via an α-methoxycarbonylation followed by a Robinson annulation sequence with methyl vinyl ketone [15]. Treatment of ester 10 with LiAlH4 in THF followed by DMP oxidation of the resultant diol (structurenot shown) provided aldehyde11with62% yieldover two steps. Selective olefination of the aldehyde carbonyl group in 11 in the presence of NaH and (MeO)2P(O)CH2CO2Me afforded the requisite E-enoate 9 (73% yield) smoothly. Thus, the stage was set for exploringthe crucial intramolecular Diels–Aldercyclo additionto generate the rigid cyclopropane moiety. Pleasingly, a sequential procedure including treatment of 9 with TBSOTf and 2, 2, 6, 6- tetramethylpiperidine (TMP) in THF at –78 ℃ (0.5 h), concentration of the reaction mixture, heating the residue in mesitylene at 150 ℃ (18 h), switchof the solvent toCH2Cl2, and desilylation using p-TsOH (2 h), furnished the desired tetracyclic keto ester 12 in one pot with 74% yield.The structure of 12 waslaterfullydeterminedby the X-ray crystallographic analysis of a derivative (vide infra). Experimental procedures and characterization data are deposited in Supporting information.

|
Download:
|
| Scheme 2. Intramolecular Diels–Alder approach to the tetracyclic intermediate 12.Reagents and conditions: (a) LiAlH4, THF; (b) DMP, CH2Cl2, 62% (2 steps); (c)(MeO)2P(O)CH2CO2Me, NaH, THF, 73%; (d) TBSOTf, TMP, THF, –78 ℃; thenmesitylene, 150 ℃; then p-TsOH, CH2Cl2, 74% (from 9). | |
{kind=link}
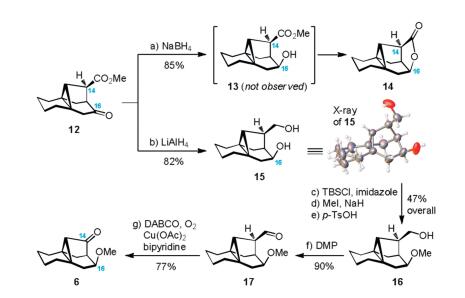
With the key intermediate 12 in hand, the remaining task was to install the requisite C14 ketone and C16-OMe functionalities (Scheme 3). Initially, reduction of the ketone carbonyl group in 12 with NaBH4 afforded lactone 14 exclusively in 85% yield. Of note, this reaction did not rest on the stage of free alcohol (13), which was prone to undergo further lactonization with the adjacent C14- ester. Facile formation of the γ-lactone indicated that reduction of the C16 ketone proceeded from the less hindered convex face, thereby permitting us to establish the C16 stereocenter with desired stereochemistry. While direct installation of the C16-OMe in the presence of C14-ester proved to be problematic, we opted to reduce both carbonyl groups in 12 with LiAlH4, leading to diol 15 with 82% yield. The structure of 15 was unambiguously verified by X-ray crystallography (Supporting information). Next, a three-step sequence including temporary masking of the primary hydroxyl group with TBSCl/imidazole, methylation of the C16 hydroxyl group with MeI/NaH, and desilylation in the presence of p-TsOH allowed the generation of compound 16 (47% overall yield from 15). Oxidation of alcohol 16 then took place in the presence of Dess-Martin periodinane (DMP) to provide aldehyde 17 (90% yield). Finally, subjecting aldehyde 17 to the oxidative deformylation conditions [DABCO, O2, Cu(OAc)2, bipyridine, DMF, 70 ℃] [16] secured the desired ketone 6 with 77% yield.

|
Download:
|
| Scheme 3. Synthesis of the 6/3/5/6 core (6) of vilmoraconitine. Reagents and conditions: (a) NaBH4, MeOH, 85%; (b) LiAlH4, THF, 82%; (c) TBSCl, imidazole, DMF, 0 ℃, 75%; (d) MeI, NaH, DMF, 69%; (e) p-TsOH, MeOH, 91%; (f) DMP, CH2Cl2, 90%; (g)DABCO, O2, Cu(OAc)2, bipyridine, DMF, 70 ℃, 77%. | |
{kind=link}
In summary, we disclosed an intramolecular Diels–Alder cycloaddition approach to the congested 6/3/5/6 tetracyclic core of the unique rearranged-type C19-diterpenoid alkaloids. Such a thermal-promoted process again proved to be useful for accessing highly substituted cyclopropane units in complex natural products [14o–q]. Application of the model studies to total synthesis of the rearranged-type C19-diterpenoid alkaloid vilmoraconitine is ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper
AcknowledgmentsFinancial support was provided by the National Natural Science Foundation of China (No. 21871189), National Science and Technology Major Projects for "Major New Drugs Innovation and Development" (No. 2018ZX09101003-005-004), and 111 Project (No. B18035). We thank Dr. Daibing Luo at the Analytical & Testing Center of Sichuan University for X-ray crystallographic structure determination.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at dio: https://doi.org/10.1016/j.cclet.2020.01.043.
| [1] |
(a) F.P. Wang, Q.H. Chen, The C19-diterpenoid alkaloids, in:G.A. Cordell (Ed.), The Alkaloids:Chemistryand Biology, 69, Elsevier Science, New York,2010,pp.1-577; (b) F.P. Wang, Q.H. Chen, X.Y. Liu, Nat. Prod. Rep. 27 (2010) 529-570. |
| [2] |
K. Wiesner, M. Götz, D.L. Simmons, et al., Tetrahedron Lett. 1 (1959) 15-24. |
| [3] |
J. Xiong, N.H. Tan, C.J. Ji, Y. Lu, N.B. Gong, Tetrahedron Lett. 49 (2008) 4851-4853. DOI:10.1016/j.tetlet.2008.06.008 |
| [4] |
(a) Y. Shen, H.L. Ai, T.W. Cao, et al., Helv. Chim. Acta 95 (2012) 509-513; (b) T.P. Yin, L. Cai, H.X. Fang, et al., Phytochemistry 116 (2015) 314-319. |
| [5] |
(a) E.C. Cherney, P.S. Baran, Isr. J. Chem. 51 (2011) 391-405; (b) A.M. Hamlin, J.K. Kisunzu, R. Sarpong, Org. Biomol. Chem. 12 (2014) 1846-1860; (c) X.Y. Liu, Y. Qin, Asian J. Org. Chem. 4 (2015) 1010-1019; (d) G. Zhu, R. Liu, B. Liu, Synthesis 47 (2015) 2691-2708; (e) X.Y. Liu, Y. Qin, Nat. Prod. Rep. 34 (2017) 1044-1050; (f) C. Dank, R. Sanichar, K.L. Choo, M. Olsen, M. Lautens, Synthesis 51 (2019)3915-3946. |
| [6] |
(a) K. Wiesner, T.Y.R. Tsai, K. Huber, S.E. Bolton, R. Vlahov, J. Am. Chem. Soc. 96(1974) 4990-4992; (b) K. Wiesner, T.Y.R. Tsai, K.P. Nambiar, Can. J. Chem. 56 (1978) 1451-1454; (c) K. Wiesner, Pure Appl. Chem. 51 (1979) 689-703. |
| [7] |
Y. Shi, J.T. Wilmot, L.U. Nordstrøm, D.S. Tan, D.Y. Gin, J. Am. Chem. Soc. 135 (2013) 14313-14320. DOI:10.1021/ja4064958 |
| [8] |
(a) C.J. Marth, G.M. Gallego, J.C. Lee, et al., Nature 528 (2015) 493-498; (b) K.G.M. Kou, S. Kulyk, C.J. Marth, et al., J. Am. Chem. Soc. 139 (2017) 13882-13896. |
| [9] |
(a) Y. Nishiyama, S. Yokoshima, T. Fukuyama, Org. Lett. 18 (2016) 2359-2362; (b) Y. Nishiyama, S. Yokoshima, T. Fukuyama, Org. Lett. 19 (2017) 5833-5835. |
| [10] |
D. Kamakura, H. Todoroki, D. Urabe, K. Hagiwara, M. Inoue, Angew. Chem. Int. Ed. 59 (2020) 479-486. DOI:10.1002/anie.201912737 |
| [11] |
(a) D. Barker, M.A. Brimble, M.D. McLeod, G.P. Savage, Org. Biomol. Chem. 2(2004) 1659-1669; (b) D.F. Taber, J.L. Liang, B. Chen, L. Cai, J. Org. Chem. 70 (2005) 8739-8742; (c) R.M. Conrad, J. Du Bois, Org. Lett. 9 (2007) 5465-5468; (d) Z.G. Liu, H. Cheng, M.J. Ge, L. Xu, F.P. Wang, Tetrahedron 69 (2013) 5431-5437; (e) H. Cheng, F.H. Zeng, D. Ma, et al., Org. Lett. 16 (2014) 2299-2301; (f) K. Hagiwara, T. Tabuchi, D. Urabe, M. Inoue, Chem. Sci. 7 (2016) 4372-4378; (g) T. Tabuchi, D. Urabe, M. Inoue, J. Org. Chem. 81 (2016) 10204-10213; (h) K. Minagawa, D. Urabe, M. Inoue, J. Antibiot. 71 (2018) 326-332; (i) Z. Lv, L. Gao, C. Cheng, et al., Chem. -Asian J. 13 (2018) 955-958; (j) M. Liu, C. Cheng, W. Xiong, et al., Org. Chem. Front. 5 (2018) 1502-1505; (k) N.A. Doering, K.G.M. Kou, K. Norseeda, et al., J. Org. Chem. 83 (2018) 12911-12920; (l) X.H. Zhou, Y. Liu, R.J. Zhou, et al., Chem. Commun. 54 (2018) 12258-12261; (m) R.J. Zhou, G.Y. Dai, X.H. Zhou, et al., Org. Chem. Front. 6 (2019) 377-382. |
| [12] |
C. Ebner, E.M. Carreira, Chem. Rev. 117 (2017) 11651-11679. DOI:10.1021/acs.chemrev.6b00798 |
| [13] |
(a) J. Gong, H. Chen, X.Y. Liu, et al., Nat. Commun. 7 (2016) 12183; (b) X.H. Li, M. Zhu, Z.X. Wang, et al., Angew. Chem. Int. Ed. 55 (2016) 15667-15671; (c) W. Nie, J. Gong, Z. Chen, et al., J. Am. Chem. Soc. 141 (2019) 9712-9718. |
| [14] |
(a) J. Zsindely, H. Schmid, Helv. Chim. Acta 51 (1968) 1510-1514; (b) T. Imagawa, M. Kawanisi, K. Sisido, J. Chem. Soc. Dalton Trans. (1971) 1292-1293; (c) P. Heimbach, K.J. Ploner, F. Thömel, Angew. Chem. Int. Ed. 10 (1971) 276-277; (d) E.N. Marvell, J. Seubert, G. Vogt, G. Zimmer, G. Moy, J.R. Siegmann,Tetrahedron 34 (1978) 1307-1322; (e) T. Imagawa, T. Nakagawa, M. Kawanisi, K. Sisido, Bull. Chem. Soc. Jpn. 52(1979) 1506-1510; (f) P. Loeliger, H. Mayer, Helv. Chim. Acta 63 (1980) 1604-1608; (g) H. Jendralla, K. Jelich, G. DeLucca, L.A. Paquette, J. Am. Chem. Soc.108 (1986)3731-3739; (h) G. Maier, N.H. Wiegand, S. Baum, et al., Chem. Ber. 122 (1989) 767-779; (i) R. ten Have, A.M. van Leusen, Tetrahedron 54 (1998) 1913-1920; (j) S.M. Ng, C.M. Beaudry, D. Trauner, Org. Lett. 5 (2003) 1701-1704; (k) J.E. Moses, J.E. Baldwin, R.M. Adlington, A.R. Cowley, R. Marquez,Tetrahedron Lett. 44 (2003) 6625-6627; (l) K.S. Khuong, C.M. Beaudry, D. Trauner, K.N. Houk, J. Am. Chem. Soc. 127(2005) 3688-3689; (m) J. Dubarle-Offner, J. Marrot, M.N. Rager, F.L. Bideau, G. Jaouen, Synlett(2007) 800-802; (n) K.C. Gue'rard, A. Gue'rinot, C. Bouchard-Aubin, et al., J. Org. Chem. 77 (2012)2121-2133; (o) C.C. Tseng, H. Ding, A. Li, Y. Guan, D.Y.K. Chen, Org. Lett. 13 (2011) 4410-4413; (p) M.J.R. Richter, M. Schneider, M. Brandstätter, E.M. Carreira, J. Am. Chem.Soc. 140 (2018) 16704-16710; (q) L.A. Wein, K. Wurst, P. Angyal, L. Weisheit, T. Magauer, J. Am. Chem. Soc.141(2019) 19589-19593. |
| [15] |
M. Boumediene, R.F. Guignard, S.Z. Zard, Tetrahedron 72 (2016) 3678-3686. DOI:10.1016/j.tet.2016.03.032 |
| [16] |
V. Van Rheenen, Tetrahedron Lett. 10 (1969) 985-988. DOI:10.1016/S0040-4039(01)97716-0 |