 2020, Vol. 31
2020, Vol. 31 


b School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
Inspired by natural photosynthesis, harvesting clean and abundant solar energy for synthetic transformations has attracted intense interests [1]. A vast number of chemists [2-5]and our group [6] have demonstrated the versatility of visible-light photocatalysts for a series of important transformations. To date, most of the activities in the field of photocatalytic synthetic transformations have still focused on the use of Ru- or r-based polypyridyl complexes, which are typically expensive and difficult to modify. Organic photoredox catalysts with the characteristics such as metal-free, low-cost, scalable and tunable, are promising alternatives because they are able to deliver unique and valuable photoredox reactivity [7]. hus, developing new and more powerful organic photoredox catalysts with both strong oxidative and reductive ability, especially those with tailorable photoredox potentials, which enable precise control of the visible-light photocatalytic reactivity, are highly desired [8].
Dehalogenation reactions are important for environmental protection [9], biochemistry [10] and organic synthesis [11] . raditional methods have often involved LiAlH4, Bu3SnH and other reducing agents [12] or noble metal catalysis, such as Ru, Pd and n catalysis [13], to form C—H bonds from C—X bonds. Recently, the generation of highly reactive aryl radicals by the photocatalytic radical reduction of aryl halides has shown great promise for C—C bond formation under mild conditions [14]. In this regard, the development of advanced organic photocatalysts to enable metalfree catalyzed C—C bond formation reactions under mild conditions utilizing inexpensive aryl halides is of significant interest.
Here, we envisaged to rationally synthesize a series of carbazolyl cyanobenzene (CCB)-based organic molecules as metal-free, photoredox potentials adjustable photocatalysts. Based on the understanding of their redox potentials, the photocatalyzed dehalogenated reduction reaction by 5CzBN as photocatalyst was developed. Meanwhile, we further applied the aryl radicals generated in situ from the photoreduction of aryl halides for C—C bond-forming reactions. A variety of arylations of pyrrole products were obtained with high selectivity and good yields under mild condition. n addition, benefiting from the mild reaction conditions, scale-up of reductive arylation of aryl halides with in continuous-flow reactor had been realized.
First, six organic photocatalysts, namely, CzPN (P1), CzPN (P2), 5CzBN (P3), 4-PAPN (P4), 3,4, 5-3CzBN (P5), and, 5-2CzPN (P6), were prepared from carbazole (diphenylamine) and fluorinated aryl nitriles by a previously reported one-step nucleophilic substitution reaction (Scheme 1) [15]. he photoredox potentials of these organic photocatalysts exhibit both strong oxidative (E1/2(*P/P-) = +1.00 ~ +1.69 V) and reductive (E1/2(P+/*P-) = -0.51 ~-1.35 V) capabilities that are comparable to many metal complexes (Tables S1 and S2 in Supporting information) and therefore are potentially suitable for reducing alkyl and aryl halides.

|
Download:
|
| Scheme 1. Structures and photoredox potentials. P1: 1, 2, 3,4-tetrakis-(carbazol-9-yl)-5, 6-dicyanobenzene (CzPN), P2: 1, 2, 3, 5-tetrakis(carbazol-9-yl)-4, 6-dicyanobenzene (CzPN), P3: 2, 3, 4, 5, 6-penta(carbazol-9-yl)benzonitrile (5CzBN), P4: 1, 2-dicyano-3,4, 5, 6-tetrakis(N, N-diphenylamino)-benzene (-PAPN), P5: 3,4, 5-tri(carbazol-9-yl)benzonitrile (3,4, 5-3CzBN), and P6: 1, 2-bis(carbazol-9-yl)-4, 5-dicyanobenzene (4, 5-2CzPN). | |
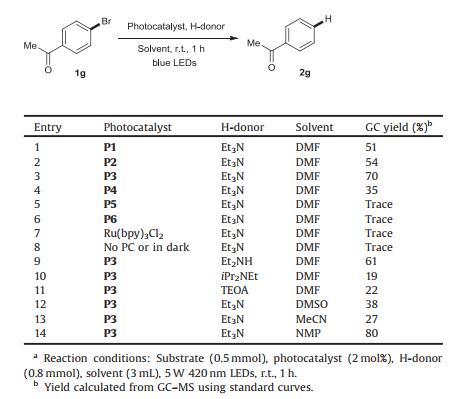
Our initial survey focused on the photocatalytic dehalogenation reaction using p-bromoacetophenone 1g as the model substrate (Table 1). As shown in able 1, the -A structure of these organic fluorophores had a major impact on their performance. Under organic photocatalysts P1 to P4 (entries 1–4), the corresponding hydrodehalogenated product was obtained successfully, and P3 furnished the highest yield (entry 3). ecreasing the number of substituted carbazole or cyano groups on the central benzene ring furnished P5 and P6, which lost their catalytic ability (entries 5 and 6). he transition metal complex Ru(bpy)3Cl2 also showed no reactivity, attesting to the advantages of organic fluorophores for this photocatalytic dehalogenation reaction (entry 7). he superior performance of P3 may be attributed to its strong reduction ability. Control experiments demonstrated the necessity of both photocatalyst and visible light (entry 8). With the screened P3 photocatalyst, we next turned our efforts towards understanding the effects of different hydrogen atom donors and solvents. The results suggested that Et3N is still the best hydrogen atom donor and that N-methyl pyrrolidone (NMP) is the optimal solvent (entries 9–14).
|
|
Table 1 Reaction condition screeninga. |
{kind=link}
Under the optimized conditions, the reaction scope was next examined with a series of aryl fluorides, aryl chlorides, aryl bromides and aryl iodines (Scheme 2). Activation of the C—and C—Cl bonds is of significant importance but is very challenging due to the high dissociation energy of these bonds. o our delight, the aryl fluorides and aryl chlorides bearing electron- withdrawing groups smoothly afforded the corresponding products 2a-2e in 46%–99% yields. Aryl bromides exhibited much better substrate versatility and functional group tolerance (1f-1m). unctional groups such as ketone (1g and 1h), cyano (1i), ester (1j), and even a very reactive aldehyde group (1f) were well compatible due to the mild conditions. An aryl-bromide bond can be selectively reduced in the presence of C—bonds (1k, 1l) because of their different reduction potentials. Notably, the reduction of 1, 3, 5-tribromobenzene 1m under the established conditions controllably produced 1, 3-dibromobenzene 2i with exceptional yield (98%). he C(sp3)-Br bond of dimethyl 2-bromomalonate 1n was also amenable to our strategy, giving the corresponding dimethyl malonate 2j in 48% yield within 10 min. inally, substituted aryl iodides, with lower reduction potentials than those of aryl bromides and aryl chlorides, gave the hydro-deiodination products in 63%–93% yields even when bearing electron-donating functionalities (2g-2m).

|
Download:
|
| Scheme 2. Scope of the hydrodehalogenation reaction. aSubstrate (0.1 mmol), 10 mol% 5CzBN was used. | |
{kind=link}
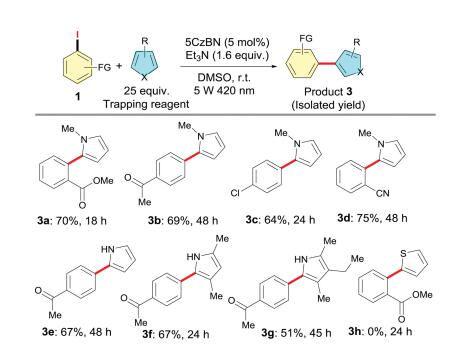
Next, we turned our efforts to utilize the aryl radicals generated in situ from the photoreduction of aryl halides for arylation reactions. N-Heterocyclic pyrroles might suit our need because of their high affinity for radical species [16]. As expected, when the reaction was carried out in the presence of an excess amount of 1- methyl-1H-pyrrole in MSO, the corresponding arylation product 3a was obtained in a yield of 70%. As shown in Scheme 3, a variety of arylation products 3a-3g were obtained at a moderate to good yields by varying the different trapping groups. unctional groups such as cyano, ester, and ketone can also be well tolerant under the mild arylation conditions. Unfortunately, when the thiophene was used to instead of pyrrole, no product 3h was observed.

|
Download:
|
| Scheme 3. Arylation of aryl iodides with substituted pyrroles. Reaction condition:substrate (0.5 mmol), trapping reagent (12.5 mmol), 5CzBN (2 mol%), Et3N(0.8 mmol), r.t., DMSO (3 mL), 5 W 420 nm LEDs. | |
{kind=link}
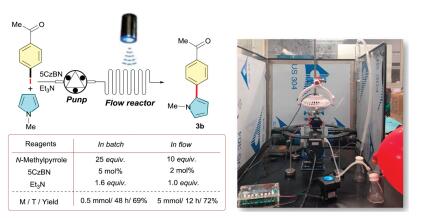
Because of the light attenuation effect through absorbing media as dictated by the Bouguer-Lambert-Beer law, the scaling up of photochemical transformations in batch is greatly limited. However, the photocatalytic continuous-flow technology shows great potential to overcome this limitation [17]. To this end, we scaled up the important arylation reactions by photocatalytic continuous-flow technology. As showed in Scheme 3, 0.5 mmol of p-bromoacetophenone were converted into target product 3b in 69% yield within 48 h under batch conditions. nterestingly, a 5 mmol scale reaction performed by continuous-flow setup was smoothly completed in much shorter time (12 h) with even less amount of catalyst and trapping reagent, affording the corresponding product 3b in 72% yield (Scheme 4).

|
Download:
|
| Scheme 4. Scale-up of arylation reaction in continuous-flow reactor. | |
{kind=link}
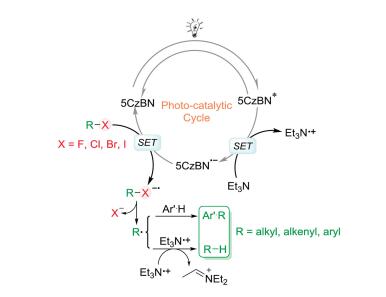
A possible mechanism is shown in Scheme 5. irst, a photoinduced single electron transfer (SE) occurs from the hydrogen donor (Et3N [E1/2 ox = 0.83 V vs. SCE]) to the excited organic photocatalyst 5CzBN (E1/2 (5CzBN*/5CzBN·-) = 1.20 V vs. SCE]), furnishing the 5CzBN radical anion with high reduction potential (E1/2(PC/PC·-) =-1.42 V vs. SCE), which reduce the active arylhalides. he SE process is well-supported by the Stern[5]–Volmer quenching studies (ig. S6 in Supporting information). he generated halogenated radical anion (R-X·-) sequentially converts to a carbon radical (R·), which either abstracts a hydrogen atom from Et3N·+ to yield the reduction product or couples with unsaturated compounds to yield the arylation product. he radical mechanism was also attested by 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (EMPO) trapped experiments as no desired product was observed [14].

|
Download:
|
| Scheme 5. Proposed catalytic mechanism of dehalogenation and arylation reactions. | |
{kind=link}
In summary, we have rationally designed and developed a series of carbazolyl cyanobenzene-based organic photoredox catalysts with tunable -A structures. he photoredox potential of the CCB-based organic photocatalysts could be fine-tuned over a broad range (‒1.35~1.69 V) by changing the number and position of carbazolyl and cyano groups on the central benzene ring. aking advantage of the broad photoredox potential, the optimized CCB-based organic photocatalyst developed herein (5CzBN) was successfully applied in both hydrodehalogenation and arylation reactions with good yields and selectivity. Studies on further applications of CCB-based photocatalysts for artificial photosynthesis of high-value-added chemicals and pharmaceuticals are currently ongoing in our laboratory.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe acknowledge the support from the National Natural Science Foundation of China (No. 21972094), Guangdong Special Support Program, Pengcheng Scholar program, Shenzhen Peacock Plan (Nos. KQJSCX20170727100802505, KQTD2016053112042971), the Educational Commission of Guangdong Province (No. 2016KTSCX126).
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at dio: https://doi.org/10.1016/j.cclet.2019.12.017.
| [1] |
(a) S. Sato, T. Morikawa, S. Saeki, T. Kajino, T. Motohiro, Angew. Chem. Int. Ed. 49(2010) 5101-5105; (b) A.D. Tjandra, J. Huang, Chin. Chem. Lett. 29(2018) 734-746; (c) C.A. Roberts, S.P. Phivilay, I.E. Wachs, Chin. Chem. Lett. 29(2018) 769-772; (d) X. Hu, S. Lu, J. Tian, et al., Appl. Catal. B 241(2019) 329-337; (e) Y. Li, Z. Yin, G. Ji, et al., Appl. Catal. B 246(2019) 12-20. |
| [2] |
D.A. Nicewicz, D.W.C. MacMillan, Science 322 (2008) 77-80. DOI:10.1126/science.1161976 |
| [3] |
M.A. Ischay, M.E. Anzovino, J. Du, T.P. Yoon, J. Am. Chem. Soc. 130 (2008) 12886-12887. DOI:10.1021/ja805387f |
| [4] |
J.M.R. Narayanam, J.W. Tucker, C.R.J. Stephenson, J. Am. Chem. Soc. 131 (2009) 8756-8757. DOI:10.1021/ja9033582 |
| [5] |
J. Liu, Q. Liu, H. Yi, et al., Angew. Chem. Int. Ed. 53 (2014) 502-506. DOI:10.1002/anie.201308614 |
| [6] |
(a) C.B. Liu, Z. Chen, C.L. Su, et al., Nat. Commun. 9(2018) 80-89; (b) C.L. Su, R. Tandiana, B.B. Tian, et al., ACS Catal. 6(2016) 3594-3599; (c) C.T. Qiu, Y.S. Xu, X. Fan, et al., Adv. Sci. 6(2019)1801403; (d) G. Zhang, W. Ou, J. Wang, et al., Appl. Catal. B 245(2019) 114-121; (e) W. Ou, G. Zhang, J. Wu, C.L. Su, ACS Catal. 9(2019) 5178-5183. |
| [7] |
(a) N.A. Romero, D.A. Nicewicz, Chem. Rev. 116(2016) 10075-10166; (b) L. Wang, M. Zhang, Y. Zhang, et al., Chin. Chem. Lett. 31(2020) 67-70. |
| [8] |
(a) T.Y. Shang, L.H. Lu, Z. Cao, et al., Chem. Commun. 55(2019) 5408-5420; (b) J. Luo, J. Zhang, ACS Catal. 6(2016) 873-877; (c) A.'. Gutierrez-Bonet, J.C. Tellis, J.K. Matsui, B.A. Vara, G.A. Molander, ACS Catal. 6(2016) 8004-8008; (d) N.R. Patel, C.B. Kelly, A.P. Siegenfeld, G.A. Molander, ACS Catal. 7(2017) 1766-1770; (e) H. Huang, X. Li, C. Yu, et al., Angew. Chem. Int. Ed. 56(2017) 1500-1505; (f) H. Huang, C. Yu, Y. Zhang, et al., J. Am. Chem. Soc. 139(2017) 9799-9802; (g) F. Wu, L. Wang, J. Chen, D.A. Nicewicz, Y. Huang, Angew. Chem. Int. Ed. 57(2018) 2174-2178; (h) J. Hou, A. Ee, H. Cao, et al., Angew. Chem. Int. Ed. 57(2018) 17220-17224; (i) C. Lévêque, L. Chenneberg, V. Corcé, C. Ollivier, L. Fensterbank, Chem. Commun. 52(2016) 9877-9880; (j) X. Tian, H. Sun, Q. Zhang, C. Adachi, Chin. Chem. Lett. 27(2016) 1445-1452; (k) W. Chen, F. Song, Chin. Chem. Lett. 30(2019) 1717-1730. |
| [9] |
T. Bhaskar, T. Matsui, J. Kaneko, et al., Green Chem. 4 (2002) 372-375. DOI:10.1039/b203745a |
| [10] |
(a) X.F. Li, M.D. Sevilla, L. Sanche, J. Am. Chem. Soc. 125(2003) 8916-8920; (b) R.L. Osborne, M.K. Coggins, J. Terner, J.H. Dawson, J. Am. Chem. Soc. 129(2007) 14838-14839. |
| [11] |
(a) M.H. Lin, W.S. Tsai, L.Z. Lin, et al., J. Org. Chem. 76(2011) 8518-8523; (b) J. Risse, M.A. Fernandez-Zumel, Y. Cudre, K. Severin, Org. Lett. 14(2012) 3060-3063; (c) M. Neumeier, D. Sampedro, M. Majek, et al., Chem. Eur. J. 24(2018) 105-108. |
| [12] |
D.Q. Yang, Z.Y. Wang, Y.H. Long, Advanced Organic Chemistry-Structure, Reaction and Mechanism, Chemical Industry Press, Beijing, 2012 p. 206.
|
| [13] |
(a) M.T. Buelow, A.J. Gellman, J. Am. Chem. Soc. 123(2001) 1440-1448; (b) N. Iranpoor, H. Firouzabadi, R. Azadi, J. Organomet. Chem. 695(2010) 887-890; (c) S. Sabater, J.A. Mata, E. Peris, Nat. Commun. 4(2013) 2553; (d) Á.F. Cañete, C.O. Salas, F.C. Zacconi, Molecules 18(2013) 398-407; (e) T. You, Z. Wang, J. Chen, Y. Xia, J. Org. Chem. 82(2017) 1340-1346. |
| [14] |
(a) A.U. Meyer, T. Slanina, C.J. Yao, B. König, ACS Catal. 6(2016) 369-375; (b) J.D. Nguyen, E.M. DAmato, J.M.R. Narayanam, C.R.J. Stephenson, Nat. Chem. 4(2012) 854-859; (c) I. Ghosh, T. Ghosh, J.I. Bardagi, B. König, Science 346(2014) 725-728; (d) C.B. Liu, Z. Chen, C.L. Su, et al., Nat. Commun. 9(2018) 80; (e) T. Maji, A. Karmakar, O. Reiser, J. Org. Chem. 76(2011) 736-739; (f) M. Neumann, S. Füldner, B. König, K. Zeitler, Angew. Chem. Int. Ed. 50(2011) 951-954; (g) J.M.R. Narayanam, J.W. Tucker, C.R.J. Stephenson, J. Am. Chem. Soc. 131(2009) 8756-8757; (h) Y.Q. Ge, P.H. Diao, X. Chen, N.N. Zhang, G. Chen, Chin. Chem. Lett. 29(2018) 903-906; (i) S.O. Poelma, G.L. Burnett, E.H. Discekici, et al., J. Org. Chem. 81(2018) 7155-7160. |
| [15] |
(a) H. Uoyama, K. Goushi, K. Shizu, H. Nomura, C. Adachi, Nature 492(2012) 234-240; (b) E. Speckmeier, T.G. Fischer, K. Zeitler, J. Am. Chem. Soc. 140(2018) 15353-15365. |
| [16] |
(a) R.A. Rossi, A.B. Pierini, A.B. Peñéñory, Chem. Rev. 103(2003) 71-168; (b) G. Sun, S. Ren, X. Zhu, M. Huang, Y. Wan, Org. Lett. 18(2016) 544-547; (c) L. Zeng, T. Liu, C. He, et al., J. Am. Chem. Soc. 138(2016) 3958-3961. |
| [17] |
(a) R. Zhou, J. Li, H.W. Cheo, et al., Chem. Sci. 10(2019) 7340-7344; (b) K. Chen, M.S. Cheung, Z. Lin, P. Li, Org. Chem. Front. 3(2016) 875-879; (c) S.M. Senaweera, A. Singh, J.D. Weaver, J. Am. Chem. Soc. 136(2014) 3002-3005; (d) U.K. Sharma, H.P.L. Gemoets, F. Schröder, T. Noel, E.V. Van der Eycken, ACS Catal. 7(2017) 3818-3823. |