 2020, Vol. 31
2020, Vol. 31 


b University of Chinese Academy of Sciences, Beijing 100049, China;
c Provincial Key Laboratory of Oil & Gas Chemical Technology, College of Chemistry and Chemical Engineering, Northeast Petroleum University, Daqing 163318, China;
d Physical Science Laboratory, Huairou National Comprehensive Science Center, Beijing 101400, China
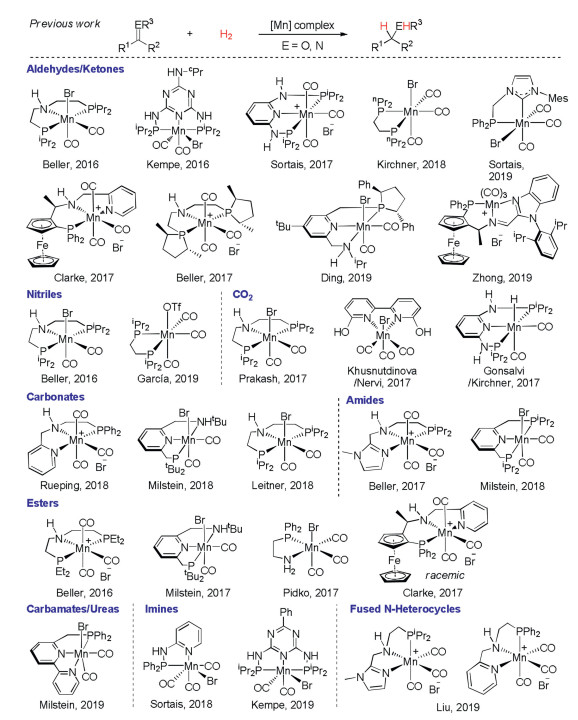
Sustainable chemistry requires precise and efficient synthesis of various functional molecules and materials in an atom- and step-economical manner. Aiming at this goal, transition metal catalysis has evolved into a powerful tool to discover new reactivity and tune chemo-, regio- and stereoselectivity for synthetic transformations. An illustrative example is transitionmetal-catalyzed hydrogenation of unsaturated molecules, which mostly has 100% atom-economy and is potentially a waste-free process [1]. Noble transition metal catalysis has played a predominant role in this field, and very high catalytic turnovers and excellent stereoselectivity have been achieved with structure-defined catalysts [2]. However, the rarity in the earth's crust and intrinsic toxicity limit future wider applications of these metals in hydrogenation reactions. As such, developments of earth abundant and less toxic 3d transition metal catalysts for hydrogenation have gained immense attention recently and are still highly desirable [3].
Manganese, as the third richest transition metal in the earth's crust, is cheap, less toxic and diverse in oxidation states (from -3 to +7) thus being a potential candidate for catalyst development [4, 5]. In this context, Beller, Kempe, Sortais, Kirchner et al. have elegantly reported hydrogenation of aldehydes/ketones by using manganese-pincer or -bidentate ligand complexes since 2016 (Scheme 1) [6]. Later on, Clarke, Beller, Ding and Zhong further developed asymmetric hydrogenation of ketones with chiral pincer-manganese catalysts [7]. Other carbonyl derivatives such as nitriles [6a, 6e, 8], carbon dioxide [9], carbonates [10], amides [11] and esters [12] could also be hydrogenated by adopting the related manganese-pincer or -bidentate ligand complexes. Very recently, Milstein and coworkers have nicely demonstrated the hydrogenation of challenging carbamates and ureas with their PNN-pincer manganese catalyst [13]. Meanwhile, Sortais, Kempe and Liu showed the hydrogenation of imines and fused N-heterocycles containing C=N bonds respectively, again with bidentate or pincertype ligand-manganese catalysts [14]. It is generally accepted that these non-innocent pincer ligands cooperate with the manganese centre to enable the hydrogenation processes through an outersphere mechanism [5]. In our continuous interest in MnH catalysis [15], we herein describe the hydrogenation of quinolines and imines by using simple manganese carbonyls, Mn2(CO)10 or MnBr(CO)5, which eliminates the previous requirement of pincer-type or bidentate ligands as well as bases (Scheme 2).During the preparation of this manuscript, Beller et al. elegantly reported a very related work using MnBr(CO)5 as a catalyst for hydrogenation of N-heterocycles at a lower temperature [16]. Of note, our results show that Mn2(CO)10 can also act as an efficient catalyst for hydrogenation of quinolines at a higher temperature. Moreover, we demonstrate that imines could be successfully hydrogenated by using simple manganese carbonyl catalysts.

|
Download:
|
| Scheme 1. Pincer or bidentate ligand-manganese complex catalyzed hydrogenation of C = E (E = O, N) containing substrates | |

|
Download:
|
| Scheme 2. Simple manganese carbonyl catalyzed hydrogenation of quinolines and imines. | |
In 2014, we reported a manganese-catalyzed [4 + 2] annulation reaction of N-H imines and alkynes with the evolution of H2 gas [15]. Mechanistic studies indicated the involvement of MnH(CO)5 as a true catalytic species in the reaction. Curious whether the simple MnH(CO)5 species could enable the hydrogenation reactions without using previous external pincer or bidentate ligands, we commenced our study with the hydrogenation of quinoline 1a by using simple Mn2(CO)10 as a catalyst (Table 1). The expected 1, 2, 3, 4-tetrahydroquinoline 2a was formed quantitatively when the hydrogenation was carried out with 5 MPa of H2 and 10 mol% of Mn2(CO)10 in THF at 150 ℃ (entry 1). While phosphine ligands were detrimental to the reaction, NPh3 and AsPh3 showed no influence on the reaction outcome (entries 2–6). Changing the reaction temperature suggested that 130 ℃ was the optimal (entries 7 and 8). Variations on the pressure of H2 showed that the reaction could proceed at lower pressure even at 1 atm, yet in decreased yields (entries 9–12). The amount of Mn2(CO)10 could be reduced to 5 mol%, however, 1 mol% of the catalyst failed in the reaction (entries 13–14). The use of MnBr bold (CO)5 instead of Mn2(CO)10 gave also a quantitative yield of 2a (entry 15).
|
|
Table 1 Optimization of reaction parameters.a |

{kind=link}
{kind=link}
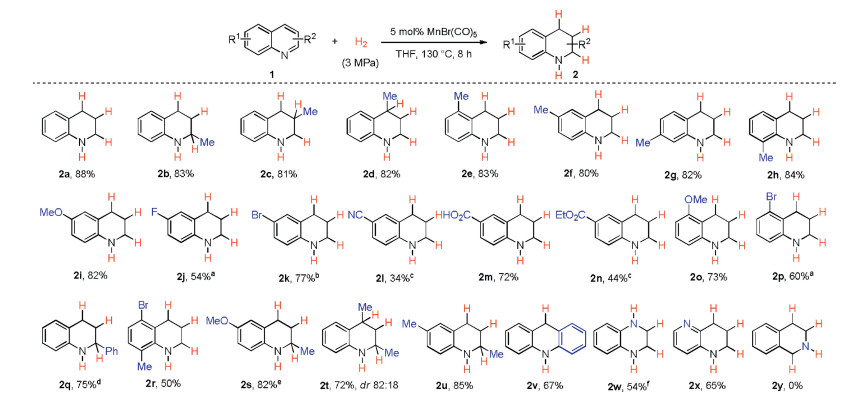
Next, the scope of quinolines was tested with the aboveobtained reaction conditions (Scheme 3). It was shown that substitutions on various positions of quinolines 1 had no obvious effect on the reaction outcome giving the corresponding 1, 2, 3, 4-tetrahydroquinolines in high yields (2a-h). Both electrondonating and -withdrawing groups were well tolerated in the reactions (2i-n). Quinolines bearing other substitution patterns were also applicable to this protocol (2o-u). The use of acridine gave 9, 10-dihydroacridine (2v) smoothly in moderate yield. Quinoxaline and 1, 5-naphthyridine led to the expected products successfully (2w, 2x) with the latter only one ring reduced (2x). Surprisingly, isoquinoline failed to afford the corresponding product (2y) under the current reaction conditions [16].

|
Download:
|
| Scheme 3. Simple manganese carbonyl catalyzed hydrogenation of quinolines. Reaction conditions: 1 (0.5 mmol), MnBr(CO)5 (5 mol%), H2 (3 MPa), 130 ℃, THF (2.5 mL), 8 h. a 24 h. b 150 ℃. c 1.0 equiv. of CH3CO2H was added. d H2 (5 MPa), 24 h. e MnBr(CO)5 (10 mol%), H2 (5 MPa), 24 h. f 12 h. | |
{kind=link}
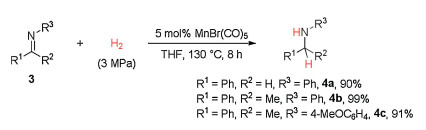
Encouraged by the above success on hydrogenation of quinoline derivatives, we further examined our catalytic system with hydrogenation of imines. As shown in Scheme 4, the hydrogenation of both aldimine and ketimines 3 could give the expected amine products 4 in excellent yields under the same reaction conditions as those of quinolines.

|
Download:
|
| Scheme 4. Simple manganese carbonyl catalyzed hydrogenation of imines. Isolated yields were shown. | |
{kind=link}
To explore the possible reaction mechanism, a set of experiments was conducted (Scheme 5). First, some additives were added to the reaction in order to probe the possible radical nature of this process. It turned out that TEMPO inhibited the reaction completely and TEMPO-H was detected by GC–MS analysis (Scheme 5a), which may result from the reaction of TEMPO with MnH(CO)5. Other radical scavengers such as butylated hydroxytoluene (BHT) and 1, 1-diphenylethylene (1, 1-DPE) affected the reaction outcome, however, the expected product 2a could still be obtained in decreased yields. To further test whether transient radical intermediates were generated in the reduction, cyclopropanyl substituted quinolines at 2-, 3- or 4-positions were subjected to the reactions and no ring-opened products were detected in all cases (Scheme 5b), which suggested the formation of a carbon radical adjacent to the cyclopropanyl group might not occur in the reaction. Remarkably, obvious deuteriumincorporation was found at 2-, 3- and 4-positions of the product with the 3-position being the most when the reaction was carried out in the presence of D2O (Scheme 5c). It indicated that MnH(CO)5 may undergo H/D exchange with D2O in the reaction. Interestingly, no D-incorporation was observed in the product when the reaction was conducted in THF-d8. To probe whether 1, 2-reduction species 5a was the possible reaction intermediate, 5a was synthesized and subjected to a series of different reaction conditions (Scheme 5d). It was shown that 5a completely transformed to product 2a under the standard reaction conditions. Interestingly, disproportionation of 5a into quinoline 1a and tetrahydroquinoline 2a took place even without MnBr(CO)5 or any external base [14c]. Such phenomenon was also found when the reactions were carried out in the absence of H2. Of note, the yields of 1a and 2a were not equal in these reactions, indicating a possible evolution of H2 during the disproportionation process.

|
Download:
|
| Scheme 5. Mechanistic studies for manganese carbonyl catalyzed hydrogenation of quinolines. Yields were determined by 1H NMR analysis. a Detected by GC–MS analysis. ND = not detected. | |
{kind=link}
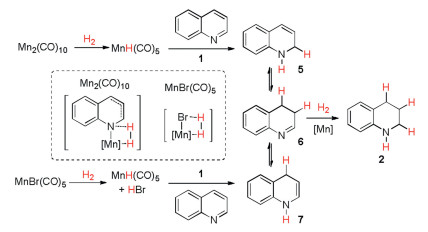
Based on the above results, we prefer an ionic MnH-promoted hydrogenation to a radical mechanism for these reactions and the tentative reaction pathways were depicted in Scheme 6. In the case of Mn2(CO)10 with H2, MnH(CO)5 was generated first and reacted with quinoline 1 through an either 1, 4-addition or 1, 2-addition way. The resulting [Mn-N] species may cleave H2 to regenerate MnH(CO)5, which could further reduce the heterocyclic intermediates 5–7 to the final tetrahydroquinoline product 2. In the case of MnBr(CO)5 with H2, MnH(CO)5 was formed together with HBr, which could activate quinoline 1 by forming a salt [16]. Two sequential steps of reduction would eventually give the tetrahydroquinoline product 2.

|
Download:
|
| Scheme 6. A proposed mechanism. | |
{kind=link}
In conclusion, we have developed a simple protocol to achieve the hydrogenation of quinoline derivatives and imines by using commercially available manganese carbonyls, Mn2(CO)10 and MnBr(CO)5, which eliminates the need of pincer-type or bidentate ligands previously commonly used. Mechanistic studies suggested the involvement of MnH(CO)5-promoted non-radical reduction process with H2. Further investigations on MnH(CO)5-enabled catalytic reduction of other substrates and detailed mechanistic studies are underway in our laboratory.
Declaration of competing interestWe herein declare that all the authors have no conflict of interest.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 21772202, 21831008), Beijing Municipal Science & Technology Commission (No. Z191100007219009) and Beijing National Laboratory for Molecular Sciences (No. BNLMS-CXXM- 201901) is gratefully acknowledged.
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.025.
| [1] |
J.G. De Vries, C.J. Elsevier, The Handbook of Homogeneous Hydrogenation[J]. Wiley-VCH.Weinheim (2007). |
| [2] |
(a) R. Noyori, T. Ohkuma, Angew. Chem. Int. Ed. 40 (2001) 40-73; (b) J.H. Xie, S.F. Zhu, Q.L. Zhou, Chem. Rev. 111 (2011) 1713-1760; (c) B. Zhao, Z. Han, K. Ding, Angew. Chem. Int. Ed. 52 (2013) 4744-4788; (d) Y.M. He, Y. Feng, Q.H. Fan, Acc. Chem. Res. 47 (2014) 2894-2906; (e) D.S. Wang, Q.A. Chen, S.M. Lu, Y.G. Zhou, Chem. Rev. 112 (2012) 2557-2590; (f) W. Tang, X. Zhang, Chem. Rev. 103 (2003) 3029-3069. |
| [3] |
(a) Z. Zhang, N.A. Butt, M. Zhou, D. Liu, W. Zhang, Chin. J. Chem. 36 (2018) 443-454; (b) G.A. Filonenko, R. van Putten, E.J.M. Hensen, E.A. Pidko, Chem. Soc. Rev. 47 (2018) 1459-1483; (c) W. Liu, B. Sahoo, K. Junge, M. Beller, Acc. Chem. Res. 51 (2018) 1858-1869; (d) W. Ai, R. Zhong, X. Liu, Q. Liu, Chem. Rev. 119 (2019) 2876-2953; (e) T. Irrgang, R. Kempe, Chem. Rev. 119 (2019) 2524-2549; (f) L. Alig, M. Fritz, S. Schneider, Chem. Rev. 119 (2019) 2681-2751. |
| [4] |
(a) D.A. Valyaev, G. Lavigne, N. Lugan, Coord. Chem. Rev. 308 (2016) 191-235; (b) W. Liu, L. Ackermann, ACS Catal. 6 (2016) 3743-3752; (c) J.R. Carney, B.R. Dillon, S.P. Thomas, Eur. J. Org. Chem. 2016 (2016) 3912-3929; (d) M. Garbe, K. Junge, M. Beller, Eur. J. Org. Chem. 30 (2017) 4344-4362; (e) R.J. Trovitch, Acc. Chem. Res. 50 (2017) 2842-2852; (f) A. Mukherjee, D. Milstein, ACS Catal. 8 (2018) 11435-11469; (g) Y. Hu, B. Zhou, C. Wang, Acc. Chem. Res. 51 (2018) 816-827; (h) X. Yang, C. Wang, Chem. Asian J. 13 (2018) 2307-2315. |
| [5] |
(a) B. Maji, M. Barman, Synthesis 49 (2017) 3377-3393; (b) F. Kallmeier, R. Kempe, Angew. Chem. Int. Ed. 57 (2018) 46-60; (c) N. Gorgas, K. Kirchner, Acc. Chem. Res. 51 (2018) 1558-1569. |
| [6] |
(a) S. Elangovan, C. Topf, S. Fischer, et al., J. Am. Chem. Soc. 138 (2016) 8809-8814; (b) F. Kallmeier, T. Irrgang, T. Dietel, R. Kempe, Angew. Chem. Int. Ed. 55 (2016) 11806-11809; (c) A. Bruneau-Voisine, D. Wang, T. Roisnel, C. Darcel, J.B. Sortais, Catal. Commun. 92 (2017) 1-4; (d) D. Wei, A. Bruneau-Voisine, T. Chauvin, et al., Adv. Synth. Catal. 360 (2018) 676-681; (e) S. Weber, B. Stoeger, K. Kirchner, Org. Lett. 20 (2018) 7212-7215; (f) M. Glatz, B. Stoger, D. Himmelbauer, L.F. Veiros, K. Kirchner, ACS Catal. 8 (2018) 4009-4016. |
| [7] |
(a) M.B. Widegren, G.J. Harkness, A.M. Slawin, D.B. Cordes, M.L. Clarke, Angew. Chem. Int. Ed. 56 (2017) 5825-5828; (b) M. Garbe, K. Junge, S. Walker, et al., Angew. Chem. Int. Ed. 56 (2017) 11237-11241; (c) M. Garbe, Z. Wei, B. Tannert, et al., Adv. Synth. Catal. 361 (2019) 1913-1920; (d)L.Zhang, Y.Tang, Z.Han, K.Ding, Angew.Chem.Int.Ed.58 (2019)4973-4977; (e) F. Ling, H. Hou, J. Chen, et al., Org. Lett. 21 (2019) 3937-3941. |
| [8] |
J.A. Garduno, J.J. García, ACS Catal. 9 (2019) 392-401. DOI:10.1021/acscatal.8b03899 |
| [9] |
(a) A. Dubey, L. Nencini, R.R. Fayzullin, C. Nervi, J.R. Khusnutdinova, ACS Catal. 7 (2017) 3864-3868; (b) F. Bertini, M. Glatz, N. Gorgas, et al., Chem. Sci. 8 (2017) 5024-5029; (c) S. Kar, A. Goeppert, J. Kothandaraman, G.K.S. Prakash, ACS Catal. 7 2017) 6347-6351. |
| [10] |
(a) A. Kumar, T. Janes, N.A. Espinosa-Jalapa, D. Milstein, Angew. Chem. Int. Ed. 57 (2018) 12076-12080; (b) V. Zubar, Y. Lebedev, L.M. Azofra, et al., Angew. Chem. Int. Ed. 57 (2018) 13439-13443; (c) A. Kaithal, M. Hoelscher, W. Leitner, Angew. Chem. Int. Ed. 57 (2018) 13449-13453. |
| [11] |
(a) V.Papa, J.R.Cabrero-Antonino, E.Alberico, etal., Chem.Sci.8 (2017) 3576-3585; (b) Y.Q. Zou, S. Chakraborty, A. Nerush, et al., ACS Catal. 8 (2018) 8014-8019. |
| [12] |
(a) S. Elangovan, M. Garbe, H. Jiao, et al., Angew. Chem. Int. Ed. 55 (2016) 15364-15368; (b) R. van Putten, E.A. Uslamin, M. Garbe, et al., Angew. Chem. Int. Ed. 56 (2017) 7531-7534; (c) N.A. Espinosa-Jalapa, A. Nerush, L.J. Shimon, et al., Chem. Eur. J. 23 (2017) 5934-5938; (d) M.B. Widegren, M.L. Clarke, Org. Lett. 20 (2018) 2654-2658. |
| [13] |
U.K. Das, A. Kumar, Y. Ben-David, M.A. Iron, D. Milstein, J. Am. Chem. Soc. 141 (2019) 12962-12966. DOI:10.1021/jacs.9b05591 |
| [14] |
(a) D. Wei, A. Bruneau-Voisine, D.A. Valyaev, N. Lugan, J.B. Sortais, Chem. Commun. 54 (2018) 4302-4305; (b) F. Freitag, T. Irrgang, R. Kempe, J. Am. Chem. Soc. 141 (2019) 11677-11685; (c) Y. Wang, L. Zhu, Z. Shao, et al., J. Am. Chem. Soc. 141 (2019) 17337-17349. |
| [15] |
(a) R. He, Z.T. Huang, Q.Y. Zheng, C. Wang, Angew. Chem. Int. Ed. 53 (2014) 4950-4953; (b) X. Yang, C. Wang, Angew. Chem. Int. Ed. 57 (2018) 923-928; (c) X. Yang, C. Wang, Chin. J. Chem. 36 (2018) 1047-1051. |
| [16] |
V. Papa, Y. Cao, A. Spannenberg, K. Junge, M. Beller, Nat. Catal. 3 (2020) 135-142. DOI:10.1038/s41929-019-0404-6 |