 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Natural Medicines, Department of Physiology, China Pharmaceutical University, Nanjing 210009, China;
c Department of Molecular and Medical Pharmacology, University of California, Los Angeles, CA 90095, United States
Ischemic stroke is characterized by high morbidity, mortality, disability and recurrence rates with serious hazard for human life and health. To search for novel drugs for safe and effective treatment of ischemic stroke, great efforts have been made; however, to date there is still a lack of ideal small molecule drug(s) [1-3].
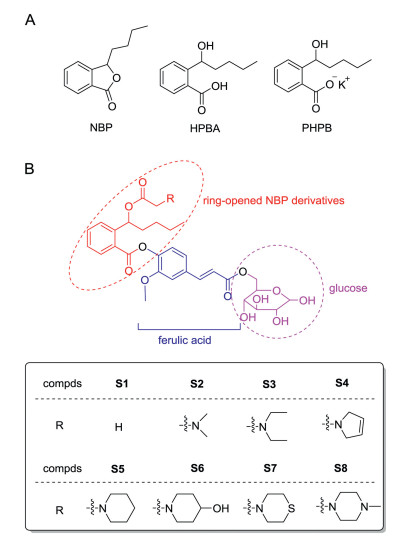
3-n-Butylphthalide (NBP) was approved in China for treatment of ischemic stroke in 2004 [4]. NBP can reduce the infarct size and improve the energy metabolism after cerebral ischemia [5, 6]. However, the therapeutic effects of NBP are moderate and it is often administered together with other drug(s) [7]. The relatively low efficacy of NBP is partly attributed to its poor aqueous solubility [8]. It has been reported that the potassium salt of 2-(1-hydroxy-n-pentyl)benzoate (PHPB), which derives from ringopening of NBP (HPBA, Fig. 1A), significantly improves aqueous solubility of NBP [9]. In addition, hybrids of the HPBA derivative with other drug/agent, such as edaravone [10], isosorbide [11], telmisartan [12], nitric oxide donors [13], etc., strengthen the therapeutic effects on acute cerebral ischemia, thus providing a principle concept of ring-opened NBP derivatives for the intervention of ischemic stroke.

|
Download:
|
| Fig. 1. (A) The chemical structures of NBP, HPBA and PHPB. (B) The design and structures of target compounds S1-S8. | |
It has been reported that oxidative stress plays an important role in cerebral ischemia/reperfusion (I/R) injury [14]. High levels of reactive oxygen species (ROS) can destroy cellular macromolecules, leading to autophagy, apoptosis and necrosis [15]. Therefore, inhibition of ROS production and/or up-regulation of endogenous antioxidant systems in vivo is one of the important strategies for designing anti-ischemic drugs. Ferulic acid (FA) is one of the main active ingredients of Chinese herbal medicines such as Angelica, Chuanxiong and Awei, and possesses a large variety of pharmacological activities, including inhibition of ROS, antioxidation, anti-inflammatory, and anti-platelet aggregation, etc. [16]. Importantly, FA can be used alone or as a linker between two active molecules to enhance the therapeutic effects of cardiacerebrovascular diseases [17, 18].
Glucose transporters 1 (GLUT1) is the most abundant transporter in blood-brain barrier (BBB) and able to deliver large amounts of aqueous glucose from peripheral circulatory system into brain, maintaining energy metabolism in the brain [19]. In recent years, GLUT1 is frequently utilized for design of braintargeted drugs [20, 21]. Fernandez et al. conjugated dopamine with glucose-1, -3 and -6 hydroxyl, respectively, to yield a series of glucose/dopamine hybrids [22]. It was found that the dopamine/ glucose-6-hydroxyl hybrid displayed the strongest BBB penetrating and antiparkinsonian activities. Mueckler et al. observed that in contrast to other the glucose hydroxyls, the 6-hydroxyl does not form a hydrogen bond that is crucial for affinity of glucose at the binding site of GLUT1, which indicates that the structural modification of the 6-hydroxyl group can retain affinity of glucose to GLUT1 [23]. In addition, the expression of GLUT1 on BBB is reportedly elevated when brain tissue is under ischemic and hypoxic conditions [24]. These investigations led us to hypothesize that glucose-6-hydroxyl derivatives of HPBA may not only improve the aqueous solubility, but also enhance the brain distribution via GLUT1-mediated BBB penetration.
To test the hypothesis, we designed and synthesized new compounds S1-S8, in which ring-opened derivatives of NBP were conjugated with glucose-6-hydroxyl by using FA as a linker (Fig. 1B). It is expected that these trihybrids may not only improve aqueous solubility owing to the hydrophilic property of glucose, but also can be recognized by GLUT1 to pass through BBB and increase concentrations of active compound(s) in the brain to exert more potent anti-ischemic activity than NBP.
The synthetic routes of target compounds S1-S8 were depicted in Scheme 1A. Given that the R- and S-NBP show almost comparable activities and NBP in market is racemic, for simplicity, we used (R/S)-NBP as starting material. (R/S)-NBP was ring-opened under basic conditions to generate hydroxyl intermediate 1, which was directly treated with acetyl chloride or chloroacetyl chloride to give esters (R/S)-2a or -2b. The two esters were then condensed with tert-butyl ferulate 10 in the presence of N, N'-dicyclohexyl carbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) to form conjugates (R/S)-3a and -3b, followed by removal of the tert-butyl group with trifluoroacetic acid (TFA) to afford acids (R/S)-4a and -4b. Subsequently, they were condensed with trimethylsilyl (TMS)- protected D-(+)-glucose 8 to offer D-(+)-glucose-6-OH esters 5a and 5b (each as epimeric mixture). The TMS groups in 5a were removed by using TFA to yield the target compound S1 as epimeric mixture. Compound 5b was reacted with dimethylamine, diethylamine, 2, 5-dihydro-1H-pyrrole, piperidine, 4-hydroxypiperidine, thiomorpholine and 1-methylpiperazine, respectively, under alkaline conditions to generate amines 6a-g. The TMS groups in 6a-g were then removed with TFA to provide the target compounds S2-S8 (each as epimeric mixture).

|
Download:
|
| Scheme 1. The synthetic route of target compounds S1-S8. (A) Reagents and conditions: (a) (1) NaOH, CH3OH, H2O, 50 ℃, 0.5 h; (2) 5% HCl (aq.), –10 ℃ to 0 ℃; (b) acetyl chloride or chloroacetyl chloride, Et3N, DMAP, DCM, –10 ℃, 5 h; (c) DCC, DMAP, DCM, 0 ℃ to r.t., 6 h; (d) CF3COOH, DCM, r.t., 3 h; (e) 8, DCC, DMAP, DCM, 0 ℃ to r.t., 8 h; (f) CF3COOH, DCM, r.t., overnight; (g) corresponding amines, Et3N, DCM, 24 h; (h) CF3COOH, DCM, r.t., overnight. (B) Reagents and conditions: (i) TMSCl, HMDS, pyridine, r.t., overnight; (j) acetone, methanol, acetic acid, 40 ℃, 4 h; (k) tert-butanol, toluene, 100 ℃; (l) vanillin, piperidine, pyridine, 10 h. | |
The intermediates 8 and 10 for synthesis of target compounds were prepared as shown in Scheme 1B. All the hydroxyl groups of D-(+)-glucose were protected by trimethylchlorosilane (TMSCl) furnishing trimethylsilyl ether 7, followed by selective deprotection in acetic acid to give 6-OH derivative 8. Moreover, Meldrum's acid (MA) was condensed with tert-butyl alcohol to form ester 9, which was treated with vanillin in the presence of a catalytic amount of pyridine and piperidine to produce tert-butyl ferulate 10.
The ratio of two epimers of the target compounds was determined by HPLC using a chiral column (CHIRALPAK® AD-H, Lot No. 19325, Daicel Chiral Technologies Co., Ltd.), exemplified by representative compound S2, and found to be approximately 1:1 as shown in Supporting information.
All target compounds with a purity of >96%, determined by high performance liquid chromatography (HPLC) analysis, were used for following experiments. The detailed synthetic procedure, in vitro and in vivo biological evaluations, and structure characterization data of the target compounds S1-S8, including 1H NMR, 13C NMR and high-resolution mass spectrometry are shown in Supporting information.
All animal experiments and animal cares were conducted in accordance with the guidelines of the Provision and General Recommendation of Chinese Experimental Animals in China. The experimental protocols were approved by the Animal Research and Care Committee of Chine Pharmaceutical University (SYXK (SU) 2016-0011).
To investigate the anti-platelet aggregation activity in vitro of the target compounds, we firstly evaluated the inhibitory effects of S1-S8 on the adenosine diphosphate (ADP)- and arachidonic acid (AA)-induced platelet aggregation in rabbit platelet rich plasma (PRP) by using Born's turbidimetric method [25]. NBP and aspirin (ASP) were used as positive controls. As shown in Table 1, all the target compounds displayed inhibitory effects to some extent. Notably, S2 was the most active in inhibition of ADP-induced platelet aggregation (IC50 = 0.08 mmol/L), 12.9-fold more potent than NBP (IC50 = 1.03 mmol/L) as well as in inhibition of AA-induced platelet aggregation (IC50 = 0.04 mmol/L), 14.3- and 3.5-fold more potent than NBP (IC50 = 0.57 mmol/L) and ASP (IC50 = 0.14 mmol/L), respectively.
|
|
Table 1 The IC50 values of compounds S1-S8 against platelet aggregation in vitro.a |

Preliminary analysis of structure-activity relationship suggests that the R2 substituted acetyl group (Scheme 1) is important to the activity of the target compounds. The amine substituted acetyls are generally more potent than the unsubstituted acetyl (S2 and S4-S6 vs. S1). Among them, the smallest substituent is the most potent (S2 vs. S3-S6). One nitrogen-containing heterocyclic substituents exert moderate activity (S4 and S5), and introduction of one hydroxyl group to the heterocyclic ring enhances the activity (S6). Surprisingly, two heteroatom-containing substituents diminish the activity (S7 and S8). Maybe they are not easy to be recognized by GLUT1 to pass through BBB or difficult to penetrate the cells' membrane. The precise structure-activity relationship should be further investigated in due course. Since S2 displayed the best inhibitory activity against ADP- or AA-induced platelet aggregation, it was selected for the following investigations.
To examine each contribution of three moieties of S2 to the anti-platelet aggregation activity, we synthesized 11a-c, which are structurally related to the three moieties (Fig. 2A and Scheme S1 in Supporting information) and examined their inhibitory activity against ADP-induced platelet aggregation. As shown in Fig. 2B, the inhibition rates of 11a, 11b, and 11c (21.32%, 18.67%, and 5.01%, respectively) were far less than that of S2 (50.76%). Next, we investigated the inhibitory activity of these compounds in combination. The inhibition rate of the three compounds in combination (31.72%) was still less than that of S2. These results suggest that the three moieties may have synergistic inhibitory effects on ADP- or AA-induced platelet aggregation.

|
Download:
|
| Fig. 2. (A) The structures of compounds 11a-c. (B) Inhibition of ADP-induced rabbit platelet aggregation by 11a-c alone or in combination in vitro. Rabbit platelet suspensions were pre-incubated with tested compounds (0.1 mmol/L) at 37 ℃ for 5 min, followed by addition of ADP (10 μmol/L). Data are expressed as the mean ±SD of each compound from six independent experiments. ***P < 0.001 vs. S2. | |
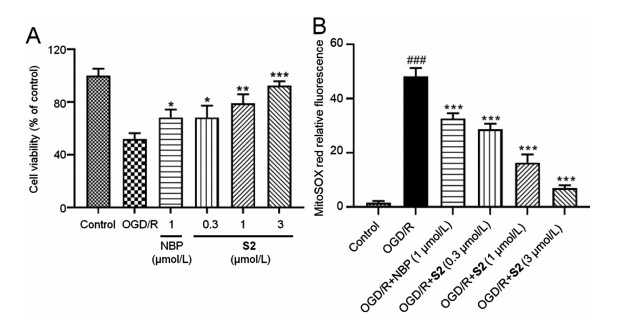
The oxygen-glucose deprivation/reoxygenation (OGD/R) model as an in vitro model for simulating I/R is commonly used for the evaluation of anti-ischemic stroke activity and mechanism investigation [26]. Thus, a primary cortical neuron cell OGD/R model (hypoxia for 2 h/reoxygenation for 24 h) was established for examining the in vitro activities of S2. As shown in Fig. 3A, compared with the control group, the neuronal cells survival rate of the OGD/R model group was significantly reduced, and S2 dosedependently increased the survival rate of neurons relative to the model group. Moreover, S2 at 1 μmol/L was more potent than NBP at the same dose. Additionally, treatment of OGD/R primary cortical neuron cells with S2 dose-dependently reduced the generation of ROS in mitochondrion (Fig. 3B). These results indicated that S2 had a significant protective effect on OGD/R-induced neuronal damage.

|
Download:
|
| Fig. 3. S2 protected primary cultured cortical neurons against OGD/R-induced injury. (A) Cell viability determined by MTT assay. (B) Mitochondrion ROS generation determined by staining with MitoSOX Red. Data are presented as mean ± SD from three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. OGD/R group; ###P < 0.001 vs. control group. | |
We then assessed the aqueous solubility of S2 at 25 ℃ by HPLC and found that S2 (16.70 ± 1.33 mg/mL) was approximately 30-fold more soluble than NBP (0.54 ± 0.21 mg/mL) in water, which is probably attributable to the water-soluble glucose and amine moieties in S2.
It is well known that BBB is a dynamic interface between the blood and brain/spinal cord with low permeability and selective ability to protect the brain from toxins and viruses in the blood circulation, but it also hinders more than 98% of small molecule agents and 100% of macromolecular agents to enter the brain tissue [27, 28]. Therefore, it is crucial for anti-cerebral ischemic drugs to penetrate BBB into brain.
The pharmacokinetics (PK) behavior of S2 in rats was firstly evaluated.Afterintravenous administration of S2 at 15 mg/kg to rats (n = 3), blood samples were taken from the suborbital vein at 0, 5, 15, 30 min and 1, 2, 4, 6, 8, 24 h and then analyzed by LC–MS/MS. The corresponding pharmacokinetic parameters of S2 are shown in Table 2. Inbrief, AUC0-∞(areaunder concentration-time curvefrom time zero to infinite), t1/2 (half-life), and Cmax (maximum plasma concentration) of S2 were 2379 ± 353 h ng mL-1, 2.75 ±0.98 h, and 6824 ± 928 ng/mL, respectively. And the CL (clearance rates) and VdSS (distribution volumes) of S2 were 105 ±15 mL min-1 kg-1 and 6.46 ± 1.9 L/kg, respectively. Moreover, the corresponding pharmacokinetic parameters of active metabolite NBP are shown in Table S1 (Supporting information), the t1/2 of NBP was up to 9.46 h, indicating that S2 possessed an improved PK behavior as compared with NBP (t1/2 = 44 min) [29].
|
|
Table 2 The plasma pharmacokinetic parameters determined after intravenous administration of S2 at a dose of 15 mg/kg to rats.a |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Furthermore, we studied the brain distribution of S2 in rats. After intravenous administration of S2 at 15 mg/kg, the rats were sacrificed at 0.5, 1 and 6 h (n = 3) and the post-processed blood and brain samples were then analyzed by LC–MS/MS. In viewof that NBP could be a main active metabolite of S2, the concentrations of both NBP and S2 were determined to calculate the brain/plasma (B/P) ratios. As shown in Table S2 (Supporting information), after intravenous administration of S2, the B/P ratios of S2 at 0.5 h and 1 h were 0.107 and 0.135, respectively. The concentration of S2 in brain at 6 h after administration was under detection limit. Interestingly, after intravenous administration of S2, the B/P ratios of NBP were significantly improved (1.99, 1.27 and 4.8 at 0.5, 1 and 6 h, respectively). Collectively, these data clearly demonstrated that S2 possessed prolonged t1/2 with higher NBP brain distributions, probably thanks to glucose-based hybridization strategy.
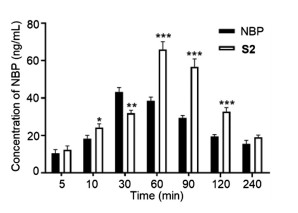
To compare the brain distribution between S2 and NBP, we conducted another assay in mice. After intravenous administration of S2 or NBP (25 mg/kg), the mice were sacrificed at 5, 10, 30, 60, 90, 120 and 240 min (n = 3), and the brain samples were then analyzed to determine the concentrations of NBP. As shown in Fig. 4, the concentrations of NBP in brain produced by S2-treatment or NBPtreatment gradually increased and then declined. And the amounts of NBP in the brain from S2 were greater than that from NBP alone in all time points except at 30 min, suggesting that S2 may more smoothly pass through BBB to release active NBP in the brain.

|
Download:
|
| Fig. 4. The concentrations of NBP in mice brain produced by treatment with S2 or NBP alone at different time. Data are expressed as mean ± SD of each group from three separate experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. NBP group. | |
{kind=link}
We next conducted the in vitro glucose competition uptake assay [30] to examine if S2 could interact with GLUT1 and be ingested into cells via it. The competitive effect of S2 with D-(+)-glucose was determined by using a cell-based assay with CellTiter-Glo® readout for ATP production in COS-7 cells in which GLUT1 is overexpressed. With the rotenone co-incubation, the cells could only produce ATP via glycolysis, thus the amount of ATP measured could be positively associated with that of glucose taken up. As shown in Fig. S1 (Supporting information), in comparison with the control group, the ATP levels was significantly reduced in a dose-dependent manner in S2 groups (10, 30, 100 μmol/L), while the NBP group (30 μmol/L) did not significantly affect the levels of ATP under the same conditions. These results suggest that S2 may be ingested via the mediation of GLUT1 to increase the NBP distribution in brain.
In summary, the trihybrids S1-S8 were designed and synthesized, inwhich the carboxylic group of a ring-opened NBP derivative hybridized with glucose 6-hydroxyl using FA as a linker. Compound S2 significantly improved aqueous solubility than NBP and displayed over 10-fold stronger inhibitory activity than NBP against ADP or AA-induced platelet aggregation. In addition, S2 was more potent than its three structural moieties alone or in combination in inhibition of platelet aggregation, suggesting that the activity of S2 may be attributed to their synergistic effects. Moreover, S2 significantlyimproved the survival rate and reducedROSgeneration of OGD/R primary neuron cells. Importantly, S2 not only possessed an improved PK profile, but also enhanced NBP distribution in rat brain, suggesting that the glucose moiety in S2 may be recognized by GLUT1, promoting S2 to pass through BBB. Collectively, our findings suggest that S2 may be a promising anti-ischemia stroke agent, which deserves further in vivo investigation.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 81773573, 81822041, 21977116 and 81673305), National Science & Technology Major Project "Key New Drug Creation and Manufacturing Program" (No. 2018ZX09711002- 006-013), the open project of State Key Laboratory of Natural Medicines (No. SKLNMZZCX201824) and State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia Fund (No. SKL-HIDCA-2018-1). Part of the work was supported by Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX18_0795).
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.02.031 .
| [1] |
V.L. Feigin, M.H. Forouzanfar, R. Krishnamurthi, et al., Lancet 383 (2014) 245-254. DOI:10.1016/S0140-6736(13)61953-4 |
| [2] |
C.J. Sommer, Acta Neuropathol. 133 (2017) 245-261. DOI:10.1007/s00401-017-1667-0 |
| [3] |
Q.W. Zhang, L. Jiang, G. Wang, J.Q. Li, Chin. Chem. Lett. 28 (2017) 1505-1508. DOI:10.1016/j.cclet.2017.02.003 |
| [4] |
X. Zhu, X. Li, J. Liu, Eur. J. Pharmacol. 500 (2004) 221-230. DOI:10.1016/j.ejphar.2004.07.027 |
| [5] |
C. Qin, P. Zhou, L. Wang, et al., J. Cereb. Blood Flow Metab. 39 (2018) 2011-2021. |
| [6] |
J. Jia, C. Wei, J. Liang, et al., Alzheimer's Dementia 12 (2016) 89-99. DOI:10.1016/j.jalz.2015.04.010 |
| [7] |
Y. Zhu, S. Tan, China J. Pract. Nerv. Dis. 13 (2010) 10-12. |
| [8] |
X. Wang, Y. Li, Q. Zhao, et al., Org. Biomol. Chem. 9 (2011) 5670-5681. DOI:10.1039/c1ob05478c |
| [9] |
J. Li, S. Xu, Y. Peng, et al., Acta Pharmacol. Sin. 39 (2018) 275-285. DOI:10.1038/aps.2017.90 |
| [10] |
X. Sheng, K. Hua, C. Yang, et al., Bioorg. Med. Chem. Lett. 25 (2015) 3535-3540. DOI:10.1016/j.bmcl.2015.06.090 |
| [11] |
X. Wang, L. Wang, T. Li, et al., J. Med. Chem. 56 (2013) 3078-3089. DOI:10.1021/jm4001693 |
| [12] |
Y. Wang, Y. Huang, Y. Xu, et al., Antioxid. Redox Signal. 28 (2018) 141-163. DOI:10.1089/ars.2017.7003 |
| [13] |
X. Wang, L. Wang, Z. Huang, et al., Bioorg. Med. Chem. Lett. 23 (2013) 1985-1988. DOI:10.1016/j.bmcl.2013.02.035 |
| [14] |
A. Chamorro, U. Dirnagl, X. Urra, A.M. Planas, Lancet Neurol. 15 (2016) 869-881. DOI:10.1016/S1474-4422(16)00114-9 |
| [15] |
R. Rodrigo, R. Fernandez-Gajardo, R. Gutierrez, et al., CNS Neurol. Disord. Drug Targets 12 (2013) 689-714. |
| [16] |
C. Cheng, T. Ho, E. Lee, et al., Am. J. Chin. Med. 36 (2008) 1105-1119. DOI:10.1142/S0192415X08006570 |
| [17] |
J. Li, Y. Zhao, G. Zhong, et al., Acta Pharm. Sin. 46 (2011) 305-310. |
| [18] |
Z. Ren, R. Zhang, Y. Li, et al., Int. J. Mol. Med. 40 (2017) 1444-1456. DOI:10.3892/ijmm.2017.3127 |
| [19] |
W.M. Pardridge, Physiol. Rev. 63 (1983) 1481-1535. DOI:10.1152/physrev.1983.63.4.1481 |
| [20] |
F. Lei, W. Fan, X.K. Li, et al., Chin. Chem. Lett. 22 (2011) 831-834. DOI:10.1016/j.cclet.2010.12.056 |
| [21] |
X.Y. Wu, X.C. Li, J. Mi, et al., Chin. Chem. Lett. 24 (2013) 117-119. DOI:10.1016/j.cclet.2013.01.022 |
| [22] |
C. Fernandez, O. Nieto, E. Rivas, et al., Carbohydr. Res. 327 (2000) 353-365. DOI:10.1016/S0008-6215(00)00073-2 |
| [23] |
M. Mueckler, C. Makepeace, J. Biol. Chem. 283 (2008) 11550-11555. DOI:10.1074/jbc.M708896200 |
| [24] |
S.J. Vannucci, L.B. Seaman, R.C. Vannucci, J. Cereb, Blood Flow Metab. 16 (19996) 77-81. |
| [25] |
G.V. Born, M.J. Cross, J. Physiol. 168 (1963) 178-195. DOI:10.1113/jphysiol.1963.sp007185 |
| [26] |
M. Agrawal, V. Kumar, A.K. Singh, et al., ACS Chem. Neurosci. 4 (2013) 285-294. DOI:10.1021/cn300143m |
| [27] |
W.M. Pardridge, NeuroRx 2 (2005) 3-14. DOI:10.1602/neurorx.2.1.3 |
| [28] |
W.M. Pardridge, Mol. Interv. 3 (2003) 90-105. DOI:10.1124/mi.3.2.90 |
| [29] |
J. Li, S.F. Xu, Y. Peng, et al., Acta Pharmacol. Sin. 39 (2018) 275-285. DOI:10.1038/aps.2017.90 |
| [30] |
H. Siebeneicher, A. Cleve, H. Rehwinkel, et al., ChemMedChem. 11 (2016) 2261-2271. DOI:10.1002/cmdc.201600276 |