 2020, Vol. 31
2020, Vol. 31 


b Hunan Provincial Key Laboratory of Materials Protection for Electric Power and Transportation, Changsha University of Science and Technology, Changsha 410114, China;
c School of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China;
d School of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China
Radical chemistry has become indispensable tools in modern organic chemistry [1]. Most of the free-radical reactions rely on transition-metal salts/ligands or environmentally-unfriendly organocatalysts, resulting in the formation of plenty of unexpected side-products and residual catalysts [2]. The disadvantages restrict the utility of these radical reactions, especially in case of preparation of artificial drugs. As a consequence, the development of simple and efficient radical reaction for clean organic synthesis is strongly desirable. Molecular oxygen is a clean, abundant and sustainable natural resource [3]. The radical process using molecular oxygen as a sole oxidant under transition metal- and organocatalysts-free conditions is undoubtedly the ideal and promising route to addressing the aforementioned challenges.
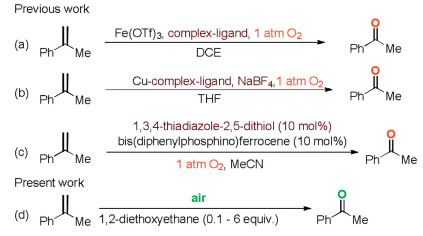
The oxidative cleavage of alkenes to corresponding carbonyl compounds is one of the most valuable transformations in organic synthesis, given the significance of these products for the fine and bulk chemical industry [4]. During the past decades, a series of protocols for the cleavage of alkenes to carbonyl compounds have been established, including Harries ozonolysis [5], (super) stoichiometric oxidation [6], transition-metal catalysis [7], and other methods [8]. Among those strategies, the transition-metalcatalyzed oxidative scission of gem-disubstituted aromatic alkenes to aryl-alkyl and diaryl ketones using eco-friendly dioxygen (1 atm) as the sole oxidant has attracted extensive attention due to its environmental benign, high efficiency, and good functional-grouptolerance. From 2015 to 2016, Xiao's group [7c] and Wang's group [7e] successively reported Fe(III) - and Cu(II)-ligand complex catalyzed oxidation of alkenes to carbonyl compounds with O2 as an oxidant (Schemes 1a and b). Very recently, Feng and He reported a bismuththiol promoted bis(diphenylphosphino)ferrocene-catalyzed oxidative scission of alkenes under ligand-free conditions (Scheme 1c) [7g]. However, these oxidation reactions have one or more of the following shortcomings, such as the noncommercially available ligands, expensive additives, and the inevitable residual transition-metal, which limits their applicability in pharmacy industry. In the other hand, the homogeneous transition-metal-catalyzed oxidations generally requires a large excess amount of volatile organic solvent as a reaction medium, which leads to a potentially flammable and possibly explosive mixture in the presence of strongly oxidative and highly energetic pure dioxygen gas [9]. In comparison with pure dioxygen gas, ambient air is a more abundant natural resource and a safer oxidant which ideally fulfils the requirements of green and sustainable chemistry [10]. To the best of our knowledge, there is no precedence of a radical oxidative scission of gem-disubstituted aromatic alkenes initiated solely by air under transition-metal-salts and additive-free conditions. Therefore, the development of efficient, safe and practical protocol for the oxidative cleavage of gem-disubstituted aromatic alkenes to carbonyl compounds is highly desirable.

|
Download:
|
| Scheme 1. Catalytic oxidative cleavage of gem-disubstituted aromatic alkenes. | |
In continuation of our efforts to develop environment benign synthetic reactions [11], herein we present a clean, safe and sustainable protocol for the oxidative cleavage of gem-disubstituted aromatic alkenes to aryl-alkyl and diaryl ketones by employing in situ generated peroxide in 6 equiv. of 1, 2-diethoxyethane under ambient air as the radical initiator (Scheme 1d).
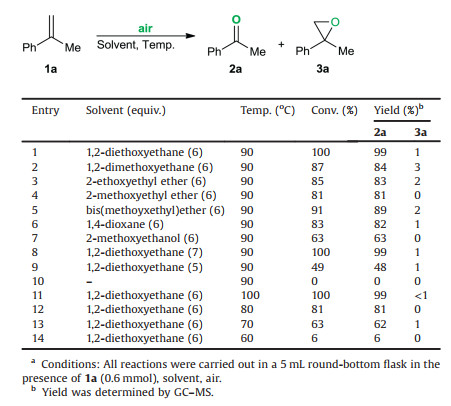
At the outset of our study, we performed the metal-free oxidation of α-menthylstyrene (1a) with ambient air in 1, 2- diethoxyethane (6 equiv.) at 90 ℃, a 99% GC–MS yield of the desired acetophenone (2a) and 1% GC–MS yield of 2-methyl-2-phenyloxirane (3a) based on 100% conversion of the starting material 1a was obtained after 12 h (Table 1, entry 1). As predicted, the nature of reaction medium exerted a significant influence on the oxidation efficiency. Compared with the slightly lower yields of 2a provided by dialkyl-end-capped ether solvents (entries 2–6), inferior reaction outcomes were obtained when monoalkyl-endcapped ethers were used as the reaction media (entry 7). The exploration on the loading of 1, 2-diethoxyathane (entries 8, 9 and 1) suggested that 6 equiv. of 1, 2-diethoxyathane was a suitable amount. No oxidation product was detected by GC–MS analysis in the absence of 1, 2-diethoxyethane (entry 10). The investigation result of the screening of reaction temperature indicated that the current oxidation could be a thermodynamically controlled process (entries 11–14). No benefit was observed by elevating the temperature to 100 ℃ (entry 11). Decreasing the reaction temperature resulted in a lower yield of 2a (entries 12 and 13) and only 6% yield of 2a was observed at 60 ℃ (entry 14).
|
|
Table 1 Optimization of reaction conditions.a |
{kind=link}
The generality of this eco-friendly oxidation reaction was then investigated under the above optimal conditions (Scheme 2). α-Methylstyreens bearing electron-neutral, -rich, or -deficient substituents at the para-, meta- or ortho-positions of the benzene ring underwent the transformation to generate the desired acetophenone derivatives with good to excellent yields (2a-2k). These experimental results suggested that neither electronic effects nor steric hindrance of α-methylstyreens has appreciable influence on the oxidation reaction outcome. Notably, these halogen substituents (F, Cl and Br) are useful entities amenable to further transformations in organic synthesis. When 4-(prop-1-en-2-yl)phenol or 4-(prop-1-en-2-yl)aniline was used as substrate, no reaction was observed. α-Methyl fused-aromatic and aromatic heterocycles can also be used as the reaction substrates and delivered the expected oxidative products (2l–2r) in good yields, thus further enhancing the scope of the developed eco-friendly oxidation reaction. It is notable that an N-heterocyclic group (1r) is well tolerated in this present oxidation reaction, but was incompatible in the previous reported transition metal-catalyzed oxidations. α-Alkylsubstituted styreens with different carbon chain lengths and isomeric structures did not significantly affect the yields of this present transformation (2s–2x), even in the presence of halogen group (2w and 2x). Other than these α-alkylsubstituted styreens, various 1, 1-diarylethylenes proved to be effective reaction substrates, producing the desired products in excellent yield under the optimal conditions (2y–2aa). Finally, substrate bearing fluorene motif also provided the desired ketone 2ab in 83% yield.

|
Download:
|
| Scheme 2. Substrate scope. All reactions were carried out in a 5 mL round-bottom flask in the presence of 1 (0.6 mmol), 1, 2-diethoxyethane (3.6 mmol), air, 90 ℃.Isolated yields are reported. | |
{kind=link}
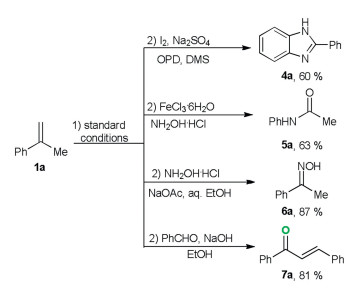
All the previous reported oxidative cleavage of aromatic alkenes requires the tedious post-processing procedures to remove the organic solvent, catalyst, additives and organic by-products, which will prevent subsequent direct chemical transformations. The present oxidation reaction is highly efficient and avoids using a large amount of volatile organic solvent, external catalyst as well as additives. In order to prove the efficiency and cleanness of the developed transformation, a series of one-pot transformations were carried out. As shown in Scheme 3, with cheap and abundant α-methylstyreen as the starting material, various valuable ketone downstream products (4a–7a) were easily obtained in good yields through one-pot oxidation/derivatization reactions [12].

|
Download:
|
| Scheme 3. One-pot transformation from 1a. | |
{kind=link}
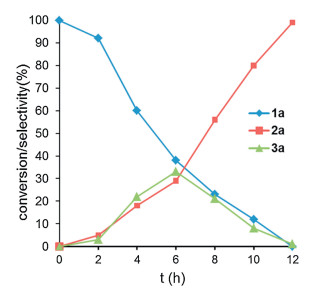
The effect of oxidation time on the conversion of α-menthylstyrene (1a) was investigated under the standard conditions and the results were shown in Fig. 1. In the initial reaction stage of 4 h, both acetophenone (2a, 16%) and 2-methyl-2-phenyloxirane (3a, 24%) were generated along with the consumption of the 1a (40% conversion), suggesting that the conversion of 1a to 7a is the dominant reaction. After reaction for 6 h, 29% yield of 2a and 34% yield of 3a based on 63% conversion of raw material 1a were detected by GC–MS. As the oxidation reaction exceeded 12 h, 1a and 3a was almost completely converted into 2a. These results suggest that the oxidation of 1a is more liable to proceed through phenyloxirane pathway, indicating a different mechanism compared with previous reports [7g].

|
Download:
|
| Fig. 1. The time course experiment for the oxidation. Conditions: 1a (0.6 mmol), 1, 2-diethoxyathane (3.6 mmol), air, 90 ℃; GC–MS yields were reported. | |
{kind=link}
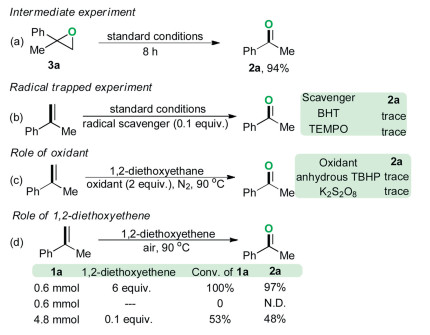
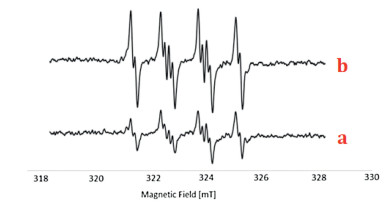
To further gain insight into the reaction mechanism, several control experiments were carried out (Scheme 4). The 2-methyl-2-phenyloxirane 3a was transformed into the acetophenone 2a in 94% GC–MS yield (Scheme 4a), suggesting that the phenyloxirane might be a key intermediate during the present oxidation reaction. Only a trace amount of oxidation product 2a was detected by GC–MS analysis in the presence of a free-radical inhibitor (BHT or TEMPO) under the optimal reaction conditions (Scheme 4c). Both the results of radical trapping experiment and radical-clock experiment [(1-cyclopropylvinyl)benzene, 1v] indicated that the present reaction might proceed through a free-radical pathway. To clarify that a free-radical species is involved in the oxidation process, the electron paramagnetic resonance (EPR) experiments were performed. As shown in Fig. 2a, when 2-diethoxyethane was exposed under oxygen atmosphere at 90 ℃ for 5 min, by using a trapping reagent of 5, 5-dimethyl-1-pyrroline N-oxide (DMPO), we successfully observed a weak signal with four characteristic hyperfines of the DMPO adduct of the peroxyl radical (DMPO-OOH) (g = 2.002, A N = 1.46 mT, AH = 1.37 mT). Prolonged the heating time to 20 min, strong signals were observed, which thereby confirmed the formation of peroxyl radicals in the reaction (Fig. 2b) [13]. The investigation on the oxidants under the nitrogen atmosphere revealed that air was pivotal in the reaction system to ensure an efficient proceeding of the metal-free oxidation reaction (Scheme 4c). A moderate yield of 2a was observed in the presence of 0.1 equiv. of 1, 2-diethoxyethane, whereas no oxidation product was detected by GC–MS analysis in the absence of 1, 2-diethoxyethane. Taken together, these experimental results manifested that the 1, 2-diethoxyethane not only served as a reaction medium but also acted as a catalyst in the developed oxidation (Scheme 4d). Considering that this type of oxidation reactions with ether as the solvent may involve the in situ generated ether peroxide, we attempted to detect this compound by GC–MS, but all efforts failed. It is probably due to the unstable of ether peroxide in high temperature. In addition, approximately 5% of 1, 2-diethoxyethylane was lost during the reaction (detected by GC–MS), which might be caused by system error.

|
Download:
|
| Scheme 4. Control experiment. | |
{kind=link}

|
Download:
|
| Fig. 2. Electron paramagnetic resonance (EPR) experiments. | |
{kind=link}
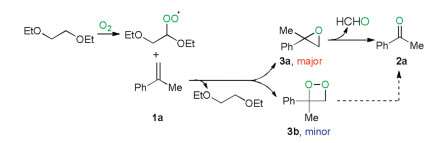
A reasonable reaction mechanism was proposed based on the above-mentioned observations and previous related reports (Scheme 5) [7g, 14]. Firstly, 1, 2-diethoxyethane reacted with dioxygen under heating conditions to produce a peroxyl radical A (detected by EPR), which oxidized α-menthylstyrene 1a to form 2-methyl-2-phenyloxirane 3a (detected by GC–MS) and regenerate the 1, 2-diethoxyethane. Finally, decomposition of 3a afforded the desired product 2a along with the regeneration of HCHO by-product. The cleavage of alkene 1 processed through dioxetane (3b) as a minor pathway cannot be completely excluded.

|
Download:
|
| Scheme 5. Plausible reaction mechanism. | |
{kind=link}
In summary, we have established an efficient and sustainable protocol for the 1, 2-diethoxyethane catalyzed oxidative cleavage of alkenes to corresponding ketones (aryl-alkyl ketones and diaryl ketones) by using ambient air as the sole oxidant. The developed methodology is compatible with reaction substrates bearing various valuable functional-groups, including heterocycles and fused ring groups. The commercially available inexpensive 1, 2-diethoxyethane plays a dual role as both a catalyst and reaction medium. Other favorable features of the present oxidation reaction are as follows: (a) Ambient air can be employed as the sole oxidant which is abundant and safe to handle; (b) The present protocol is ideally suited for one-pot sequential synthesis, owing to its clean reaction conditions, minimal amount of solvent and high to quantitative yields.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentWe are grateful for financial support from the Hunan Provincial Natural Science Foundation of China (Nos. 2019JJ20008 and 2019JJ40090).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi: https://doi.org/10.1016/j.cclet.2020.01.036 .
| [1] |
(a) H. Yi, G. Zhang, H. Wang, et al., Chem. Rev. 117 (2017) 9016-9085; (b) M.H. Huang, W.J. Hao, G. Li, S.J. Tu, B. Jiang, Chem. Commun. (Camb.) 54 (2018) 10791-10811; (c) X. Gong, G. Li, Z. Gan, et al., Asian J. Org. Chem. 8 (2019) 1472-1478; (d) Y. Zhang, K. Sun, Q. Lv, et al., Chin. Chem. Lett. 30 (2019) 1361-1368; (e) H. Yue, P. Bo, B.L. Wang, et al., Chin. J. Org. Chem. 39 (2019) 463-468; (f) G.H. Li, D.Q. Dong, Q. Deng, S.Q. Yan, Z.L. Wang, Synthesis 51 (2019) 3313-3319; (g) Q. Huang, L. Zhu, D. Yi, X. Zhao, W. Wei, Chin. Chem. Lett. 31 (2020) 373-376; (h) K. Sun, X.L. Chen, Y.L. Zhang, et al., Chem. Commun. (Camb.) 55 (2019) 12615-12618; (i) L. Wang, M. Zhang, Y. Zhang, et al., Chin. Chem. Lett. 31 (2020) 67-70; (j) G.H. Li, D.Q. Dong, X.Y. Yu, Z.L. Wang, New J. Chem. 43 (2019) 1667-1670; (k) K. Sun, Y.F. Si, X.L. Chen, et al., Adv. Synth. Catal. 361 (2019) 4483-4488; (l) L. Wang, Y. Zhang, M. Zhang, et al., Tetrahedron Lett. 60 (2019) 1845-1848; (m) S. Liu, K. Chen, W.J. Hao, et al., J. Org. Chem. 84 (2019) 1964-1971; (n) K. Sun, B. Luan, Z. Liu, et al., Org. Biomol. Chem. 17 (2019) 4208-4211; (o) L. Xiong, H. Hu, C.W. Wei, B. Yu, Eur. J. Org. Chem. 2020 (2020) 1588-1597. |
| [2] |
C. Zhang, C. Tang, N. Jiao, Chem. Soc. Rev. 41 (2012) 3464-3484. DOI:10.1039/c2cs15323h |
| [3] |
(a) X. Ji, D. Li, X. Zhou, H. Huang, G.J. Deng, Green Chem. 19 (2017) 619-622; (b) D.F. Jiang, J.Y. Hu, W.J. Hao, et al., Org. Chem. Front. 5 (2018) 189-196; (c) X. Chen, Z. Wang, H. Huang, G.J. Deng, Adv. Synth. Catal. 360 (2018) 4017-4022; (d) F.L. Zeng, X.L. Chen, S.Q. He, et al., Org. Chem. Front. 6 (2019) 1476-1480; (e) Y. Xia, H. Huang, F. Zhang, G.J. Deng, Org. Lett. 21 (2019) 7489-7492; (f) Z. Gan, Q. Yan, G. Li, et al., Adv. Synth. Catal. 361 (2019) 4558-4567; (g) X. Ji, M. Tan, M. Fu, G.J. Deng, H. Huang, Org. Biomol. Chem.17 (2019) 4979-4983; (h) K. Chen, W.J. Hao, S.J. Tu, B. Jiang, Green Chem. 21 (2019) 675-683; (i) X.Zhang, S.Dong, Q.Ding, X.Fan, G.Zhang, Chin.Chem.Lett.30 (2019) 375-378; (j) K.J. Liu, T.Y. Zeng, J.L. Zeng, et al., Chin. Chem. Lett. 30 (2019) 2304-2308; (k) K.J. Liu, J.H. Deng, J. Yang, et al., Green Chem. 22 (2020) 433-438; (l) W.M. He, Y.W. Lin, D. Yu, Sci. China Chem. 63 (2020) 291-293. |
| [4] |
(a) J.P. Wan, Y. Gao, L. Wei, Chem. Asian J. 11 (2016) 2092-2102; (b) G. Urgoitia, R. SanMartin, M.T. Herrero, E. DomÃnguez, ACS Catal. 7 (2017) 3050-3060. |
| [5] |
T.J. Fisher, P.H. Dussault, Tetrahedron 73 (2017) 4233-4258. DOI:10.1016/j.tet.2017.03.039 |
| [6] |
(a) K. Miyamoto, N. Tada, M. Ochiai, J. Am. Chem. Soc. 129 (2007) 2772-2773; (b) F.V. Singh, H.M.S. Milagre, M.N. Eberlin, H.A. Stefani, Tetrahedron Lett. 50 (2009) 2312-2316; (c) A. Rajagopalan, M. Lara, W. Kroutil, Adv. Synth. Catal. 355 (2013) 3321-3335; (d) X. Zeng, D. Xu, C. Miao, C. Xia, W. Sun, RSC Adv. 4 (2014) 46494-46497; (e) D.J. Lippincott, P.J. Trejo-Soto, F. Gallou, B.H. Lipshutz, Org. Lett. 20 (2018) 5094-5097. |
| [7] |
(a) M.M. Hossain, W.K. Huang, H.J. Chen, P.H. Wang, S.G. Shyu, Green Chem. 16 (2014) 3013-3017; (b) C. Mi, X.G. Meng, X.H. Liao, X. Peng, RSC Adv. 5 (2015) 69487-69492; (c) A. Gonzalez-de-Castro, J. Xiao, J. Am. Chem. Soc. 137 (2015) 8206-8218; (d) G. Urgoitia, R. SanMartin, M.T. Herrero, E. DomÃnguez, Adv. Synth. Catal. 358 (2016) 1150-1156; (e) Y. Liu, D. Xue, C. Li, J. Xiao, C. Wang, Catal. Sci. Technol. 7 (2017) 5510-5514; (f) C.A. Hone, A. OâorgTM Kearney-McMullan, R. Munday, C.O. Kappe, ChemCatChem 9 (2017) 3298-3302; (g) C. Zhu, D. Wei, Y. Wu, et al., J. Alloys. Compd. 778 (2019) 731-740; (h) B. Xiong, X. Zeng, S. Geng, et al., Green Chem. 20 (2018) 4521-4527. |
| [8] |
(a) T. Wang, N. Jiao, J. Am. Chem. Soc. 135 (2013) 11692-11695; (b) G.Z. Wang, X.L. Li, J.J. Dai, H.J. Xu, J. Org. Chem. 79 (2014) 7220-7225; (c) Y. Imada, Y. Okada, K. Noguchi, K. Chiba, Angew. Chem. Int. Ed. 58 (2019) 125-129; (d) Z. Cheng, W. Jin, C. Liu, Org. Chem. Front. 6 (2019) 841-845. |
| [9] |
P.M. Osterberg, J.K. Niemeierc, C.J. Welch, et al., Org. Process Res. Dev. 19 (2015) 1537-1543. DOI:10.1021/op500328f |
| [10] |
(a) X. Wang, C. Wang, Y. Liu, J. Xiao, Green Chem. 18 (2016) 4605-4610; (b) B. Liu, F. Jin, T. Wang, X. Yuan, W. Han, Angew. Chem. Int. Ed. 56 (2017) 12712-12717; (c) S. Li, B. Zhu, R. Lee, B. Qiao, Z. Jiang, Org. Chem. Front. 5 (2018) 380-385; (d) R. Li, X. Chen, S. Wei, et al., Adv. Synth. Catal. 360 (2018) 4807-4813; (e) B. Liu, P. Hu, F. Xu, et al., Comm. Chem. 2 (2019) 5-12; (f) D.Q. Dong, L.X. Li, G.H. Li, et al., Chin. J. Catal. 40 (2019) 1494-1498; (g) L.Y. Xie, J.L. Hu, Y.X. Song, et al., ACS Sustainable Chem. Eng. 7 (2019) 19993-19999; (h) L.Y. Xie, Y.S. Lu, H.R. Ding, et al., Chinese J. Catal. 41 (2020) 1168-1173; (i) G. Li, Q. Yan, X. Gong, X. Dou, D. Yang, ACS Sustainable Chem. Eng. 7 (2019) 14009-14015; (j) L.Y. Xie, Y.L. Chen, L. Qin, et al., Org. Chem. Front. 6 (2019) 3950-3955; (k) Z. Wang, X. Ji, J. Zhao, H. Huang, Green Chem. 21 (2019) 5512-5516; (l) Y. Lv, P. Bao, H. Yue, J.S. Li, W. Wei, Green Chem. 21 (2019) 6051-6055. |
| [11] |
(a) L.H. Lu, Z. Wang, W. Xia, et al., Chin. Chem. Lett. 30 (2019) 1237-1240; (b) L. Peng, Z. Hu, Q. Lu, et al., Chin. Chem. Lett. 30 (2019) 2151-2156; (c) L. Peng, Z. Hu, Z. Tang, Y. Jiao, X. Xu, Chin. Chem. Lett. 30 (2019) 1481-1487; (d) W.-H. Bao, Z. Wang, X. Tang, et al., Chin. Chem. Lett. 30 (2019) 2259-2262; (e) Z. Cao, Q. Zhu, Y.W. Lin, W.M. He, Chin. Chem. Lett. 30 (2019) 2132-2138; (f) S. Peng, Y.X. Song, J.Y. He, et al., Chin. Chem. Lett. 30 (2019) 2287-2290; (g) Z. Wang, W.M. He, Chin. J. Org. Chem. 39 (2019) 3594-3595; (h) Q.W. Gui, X. He, W. Wang, et al., Green Chem. 22 (2020) 118-122; (j) S. Peng, Y.W. Lin, W.M. He, Chin. J. Org. Chem. 40 (2020) 541-542; (i) S. Peng, D. Hu, J.L. Hu, et al., Adv. Synth. Catal. 361 (2019) 5721-5726. |
| [12] |
(a) S. Mahajan, B. Sharma, K.K. Kapoor, Tetrahedron Lett. 56 (2015) 1915-1918; (b) O. Ravi, A. Shaikh, A. Upare, K.K. Singarapu, S.R. Bathula, J. Org. Chem. 82 (2017) 4422-4428; (c) G.C. Senadi, M.R. Mutra, T.Y. Lu, J.J. Wang, Green Chem. 19 (2017) 4272-4277; (d) R. Bashary, G.L. Khatik, Bioorg. Chem. 82 (2019) 156-162. |
| [13] |
(a) G.R. Buettner, Free Radical Biol. Med. 3(1987) 259-303; (b) J. Van Der Zee, D.P. Barr, R.P. Mason, Free Radical Biol. Med. 20 (1996) 199-206. |
| [14] |
(a) M.M. Hossain, S.G. Shyu, Tetrahedron 70(2014) 251-255; (b) K.J. Liu, S. Jiang, L.H. Lu, et al., Green Chem. 20 (2018) 3038-3043. |