 2020, Vol. 31
2020, Vol. 31 


Because of its unique properties in killing tumor cells and good safety profile, tumor necrosis factor (TNF)-related apoptosisinducing ligand (TRAIL) as one of the TNF superfamily members has attracted great attentions in cancer treatment [1-3]. It can specifically bind to death receptors (DR4 and DR5) on the membrane of tumor cells to activate TRAIL-DR4/5-FADD-Caspase-8/10 signaling pathways and then initiate mitochondriadependent and -independent apoptosis of tumor cells [4, 5]. Recent years, numbers of clinical trials (NCT01088347, NCT01258608, NCT03082209, etc.) have been done or are undergoing to develop highly effective TRAIL agonist-based combination cancer treatments. However, there are still no exciting outcomes in those clinical trials because of the TRAIL resistance of most primary tumors [1, 6-8]. Therefore, developing an effective agent or strategy to enhance the sensitivity of tumor cells to TRAIL-mediated apoptosis will be greatly useful for addressing above mentioned problem. Accumulating evidences also confirmed that TRAIL agonist-based combination cancer treatment exhibited better therapeutic efficacy than monotherapy [9-11].
It is known that endoplasmic reticulum (ER) stress responses play crucial roles in the adaptation of tumor cells to hostile microenvironments in tumors like poor nutrient availability, hypoxia and oxidative stress [12]. In addition, some evidences demonstrated that ER stress could greatly enhance the sensitivity of tumor cells toTRAIL [13-16]. Our previous studies demonstrated that selenium-containing ruthenium complex could more effectively enhance the radio-sensitivity of tumor cells than that of other ruthenium complexes without selenium through activating ROS-mediated signaling pathways [17]. Meanwhile, when introduced into iron(Ⅱ) complexes containing 1, 10-phenanthroline (phen) derivatives, selenium could enhance the anti-angiogenesis activity of the complex [18]. Furthermore, selenium compounds also exhibited anticancer activities [19]. Additionally, it should be noted that some selenoproteins play important roles in ER stress [20-22].
Inspired by the unique properties of selenium-based complexes, we hypothesize that Se/Fe complex may be an effective agent facilitating TRAIL-mediated apoptosis for cancer treatment. Herein, we successfully synthesized Se/Fe complex with high activities in inhibiting tumor cells proliferation and migration capabilities through down-regulating ER stress related selenoproteins while exhibiting low toxicity to normal cells. Meanwhile, it could more efficiently destroy tumor spheroids with good penetration capability. As expected, the developed complex also could facilitate TRAIL treatment to induce tumor cells apoptosis in extrinsic and intrinsic signaling pathways via promoting robust ROS generation and down-regulating ER stress related selenoproteins. This study may shed light for the development of TRAIL-based combination cancer treatment.
According to the protocol of our previous study [18], we first synthesized the Se/Fe complex (Fig. 1a). Then, the effects of the complex on the viabilities of different cervical cancer cells (Caski, SiHa, HeLa) were evaluated using MTT assay. Interestingly, it was shown that the IC50 values of Se/Fe complex to three kinds of cancer cells were much lower than that of Fe3 complex (Fig. 1b). That is to say, Se introduction could greatly enhance the inhibition activity of Fe complex. UV–vis analysis confirmed that the higher cellular uptake of Se/Fe complex into HeLa cells might be the major contributor for its better inhibition activity than that of Fe3 complex (Fig. 1c). As demonstrated in our previous study [18], the enhanced solubility of Fe complex after Se substitution endows the better cellular uptake of Se/Fe complex. Meanwhile, distinguished from Fe3 complex with high toxicity to normal cells, Se/Fe complex exhibited relatively low toxicity at the same concentration of Fe (Figs. 1d and e). All these evidences indicated that our developed Se/Fe complex with good safety issue could highly effectively inhibit cancer cells proliferation. Given the low sensitivity of HeLa cells to TRAIL (Fig. S1 in Supporting information), we selected HeLa cells as the model in subsequent studies to explore the inhibition activity of Se/Fe complex and its underlying mechanisms.

|
Download:
|
| Fig. 1. Cytotoxicity of Se/Fe and Fe3 complexes to cervical cancer cells and normal cell lines. (a) Scheme of potent anticancer activities induced by the co-treatment of Fe complexes and TRAIL. (B) Cytotoxic effects of Fe complexes on the cervical cancer cells (Caski, SiHa, HeLa). The cervical cancer cells were exposed to Se/Fe or Fe3 with certain concentration at 37 ℃ for 24 h. (c) Cellular uptake of Fe complexes in HeLa cells. Fe complexes (32 μmol/L) were added into HeLa cells for 24 h. (d, e) Toxicity of Fe complexes to normal cell lines (L02, Chem-5). The normal cells were dealt with Fe complexes at various concentrations for 48 h. Asterisk with ** represents statistical difference at the P < 0.01 level. | |
{kind=link}
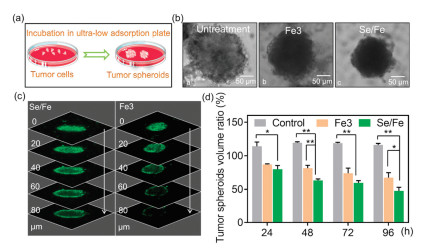
As the most often used model, multicellular tumor spheroids (MCTS) can simulate the pathophysiological conditions of tumor tissue including hypoxic areas and proliferation gradients [23-26]. Therefore, we subsequently explored the antitumor effects of the complexes using HeLa tumor spheroid model (Fig. 2a). Intriguingly, it was observed that Se/Fe complex could more efficiently induce the size reduction and morphology change of the spheroids especially when treated for a long time (i.e., over 48 h) than that of Fe3 complex (Fig. 2b). By taking full advantages of the fluorescent property of Fe3 and Se/Fe complexes [27], we carefully evaluated their penetration capabilities into tumor spheroids by CLSM. As shown in Fig. 2c, Se/Fe complex could more efficiently penetrate into deep regions of the spheroids than Fe3 complex even just treated for 12 h. Moreover, the CLSM software analysis confirmed that Se/Fe complex could more efficiently damage the tumor spheroids than that of Fe3 complex. It was shown that the volume ratio of tumor spheroids when treated with Se/Fe complex for 96 h decreased by 69% of that of untreated spheroids while Fe3 complex treatment leading to 49% reduction (Fig. 2d). All these solid evidences indicate that Se introduction into Fe complex can endow the complex with excellent inhibition activity through greatly enhancing its penetration capability and cellular uptake.

|
Download:
|
| Fig. 2. Inhibitory effect and penetration capacity of Se/Fe and Fe3 complexes towards HeLa tumor spheroid. (a) HeLa tumor spheroids model. (b) Optical imaging of HeLa tumor spheroids before/after treatment with Fe complexes for 48 h. (c) Penetration capacity analysis of Fe complexes towards HeLa tumor spheroids. The spheroids were incubated with Se/Fe or Fe3 complexes for 12 h. The tumorscanning fluorescent microscope were taken every 20 μm section from the top to the middle of tumor spheres. (d) Inhibitory effect of Fe complexes towards HeLa tumor spheroids. The complexes were used at the concentration of 32 μmol/L. Asterisks (*/**) denote significantly different P < 0.05 level and P < 0.01 level respectively. | |
{kind=link}
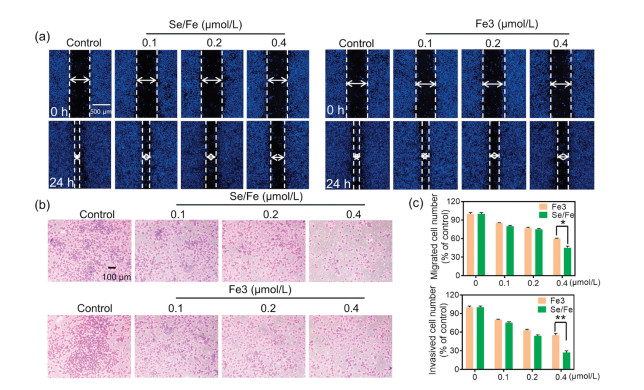
It is well known that the migration and invasion abilities of tumor cells play crucial roles in tumor progressions [28]. Besides cytotoxic effects, some metal complexes at non-toxic concentrations are also capable to effectively inhibit tumor cells metastasis [29]. Therefore, we subsequently evaluated the effects of complexes on the migration and invasion capabilities of HeLa cells using wound-healing and transwell assays. Firstly, we confirmed the non-toxic concentrations of the complexes using MTT assay. It was found that both complexes exhibited no apparent toxicity to HeLa cells at the concentrations of 0.1–0.4 μmol/L after 24 h treatment (Fig. S2 in Supporting information). Intriguingly, it was observed that Se/Fe complex could more efficiently inhibit the migration of HeLa cells than Fe3 complex in a dose-dependent manner (Figs. 3a and c). Furthermore, the data of transwell assay demonstrated that Se/Fe complex could also more effectively reduce the invasion activity of HeLa cells especially at 0.4 μmol/L than Fe3 complex (Figs. 3b and c).

|
Download:
|
| Fig. 3. The effects of Fe complexes on the migration and invasion capabilities of HeLa cells. (a) Anti-migration assay of Fe complexes on HeLa cells. (b) Anti-invasion assay of Fe complexes on HeLa cells. (c) The migration and invasion of HeLa cells in quantitative analysis with the treatment of Fe complexes. * and ** represent statistically different at the P < 0.05 level and P < 0.01 level respectively. | |
{kind=link}
As an essential element, selenium (Se) incorporated into proteins (selenoprotein) plays crucial roles for the health of humans, animals and some other microorganisms [30, 31]. To fully understand its interactions with tumor cells, we then explored the effects of both complexes on the expressions of some selenoproteins in HeLa cells using qPCR assay and western blotting analysis. From quantitative PCR analysis, it was demonstrated that Se/Fe complex could more effectively down-regulate the expressions of SELS, GPX4, SELO in HeLa cells when treated for 12 h while Fe3 complex just slightly down-regulating GPX4 and SELO (Fig. 4a). The higher cellular uptake of Se/Fe complex and its possible metabolisms into selenium-containing amino acids which may interfere with the synthesis of selenoproteins endow Se/Fe complex with greater effects on selenoproteins than that of Fe3 complex. In addition, western blotting analysis indicated that Se/Fe complex could more significantly induce the down-regulation of those three selenoproteins when cells treated for 24 h compared to untreated group (Fig. 4b). However, it was found that Fe3 complex showed no effect on GPX4 expression and up-regulated SelO expression (Fig. 4b), which are inconsistent with that of PCR analysis. These phenomena may be attributed to the different time points of those two assays and the complicated interactions among selenoproteins. The detailed underlying mechanisms still need further studies. It is known that SELS locating in endoplasmic reticulum affects the ER-associated degradation of misfolded protein [32]. SelS silence enhanced endoplasmic reticulum (ER) stress induced by hydrogen peroxide or tunicamycin to cause cell apoptosis [33]. GPX4 as one of selenocysteine-containing glutathione peroxidases exhibited anti-apoptotic capability through reducing damaging ROS [34-36]. Meanwhile, SELO located in mitochondria with C-X-X-U (where C is cytosine, X is any nucleotide, and U is uridine) motif could serve as redox-active mitochondrial selenoprotein for redox control [37]. All these evidences indicated that our developed Se/Fe complex could shape the intracellular redox state and induce ER stress to damage tumor cells (HeLa) via inhibiting the expressions of related selenoproteins.

|
Download:
|
| Fig. 4. Effects of Fe complexes on relative levels of selenoproteins. (a) The quantitative analysis on the relative RNA levels of selenoprotein genes (SELS, GPX4, SELO) in HeLa cells treated with Se/Fe or Fe3 (4 μmol/L) for 12 h. * represent statistically different at the P < 0.05 level. (b) The effects of Se/Fe or Fe3 (4 μmol/L) treatment for 24 h on the expression levels of selenoproteins (SELS, GPX4, SELO) in HeLa cells analyzed by western blotting assay. | |
{kind=link}
It is known that the resistance of tumor cells against TRAIL greatly limits the therapeutic efficacy of TRAIL-based cancer treatment [1]. Recently, accumulating evidences indicated that ER stress could sensitize tumor cells to TRAIL through downregulation of FLIP and Mcl-1 and PERK-dependent up-regulation of TRAIL-R2 [13, 15, 38]. Inspired by the unique property of Se/Fe complex in modulating ER stress related selenoproteins, we subsequently investigated whether our developed complex could improve the efficacy of TRAIL treatment. Interestingly, pretreating HeLa cells with Se/Fe complex at highest concentration (4 μmol/L) could more effectively enhance the efficacy of TRAIL agonist treatment (cell viability decreased by 42.1%) than pretreatment with Fe3 complex (cell viability decreased by 25.6%, Fig. 5a). To ascertain the reciprocity between the complexes and TRAIL, the growth suppression of single and combined treatments was assessed by isobologram analysis. As shown in Figs. 5b and c, the anti-proliferation efficacy of the associated Se/Fe complex and TRAIL treatment in the manner of different ratios (1:40) were statistically more synergistic than that of the Fe3 and TRAIL cotreatment, as verified by the fact that the position of the data points in the isometric map was significantly lower than other treatments. Furthermore, the combination index (CI) of Se/Fe complex and TRAIL co-treatment was significantly lower than that of Fe3 and TRAIL co-treatment (Fig. 5d). More importantly, both single and combination treatments (Se/Fe, Fe3, TRAIL, Se/ Fe + TRAIL, Fe3 + TRAIL) did not show any toxicity to normal cells (L02 and Chem5, Figs. S3a and b in Supporting information). Additionally, the cell cycle analysis demonstrated that Se/Fe complex at low concentration (2 μmol/L) combined TRAIL treatment significantly induced G0/G1 phase arrest and sub-G1 accumulation while Se/Fe complex and TRAIL treatment alone just slightly inducing S phase and G0/G1 phase arrest, respectively. Intriguingly, Se/Fe complex with high concentration (4 μmol/L) combination treatment mainly lead to large accumulation of subG1 population up to 31.8% (Fig. 5e and Fig. S4 in Supporting information), indicating robust cell apoptosis. It is known that reactive oxygen species (ROS) plays critical roles in inducing cell apoptosis and cell cycle arrest [39-43]. Encouraged by the good inhibition capabilities of combination treatment, we then explored the intracellular ROS level after treatment using dihydroethidium (DHE) fluorescence assay. It was found that the combination treatment induced robust generation of ROS within cells while single treatment alone just slightly enhancing the ROS generation (Fig. 5f and Fig. S5 in Supporting information). From above mentioned evidences, it can be concluded that the robust ROS generation by combination treatment together with the enhanced sensitivities of HeLa cells to ER stress and ROS-induced apoptosis by Se/Fe complex which down-regulating the expressions of SELS, GPX4 and SELO lead to the greater cytotoxicity of combination modality to TRAIL-resistant tumor cells.

|
Download:
|
| Fig. 5. The synergistic effects of Se/Fe complex with TRAIL treatment. (a) HeLa cells were treated with certain concentrations (1, 2 and 4 μmol/L) of Se/Fe or Fe3 for 24 h followed by160 ng/mL TRAIL for another 24 h. (b, c) The sensitization of Fe complexes for TRAIL on HeLa cells was determined by the isobologram analysis. The actual IC50 values of Fe complexes and TRAIL at specific proportion of concentrations in the combined therapy were represented as the data points A (1:80) and B (1:40) in the isobologram. Se/Fe or Fe3 (mmol/L):TRAIL (ng/mL) = 1:80, 1:40. (d) CI representing the specific proportion of concentrations in the combined therapy (Se/Fe or Fe3 and TRAIL). (e) The elevated sub-diploid peaks in HeLa cells after the treatment of Se/Fe (2, 4 μmol/L) followed by TRAIL (160 ng/mL) were quantitated by flow cytometric analysis. (f) The level of intracellular ROS generation in HeLa cells triggered by Se/Fe (4 μmol/L) or/and TRAIL (160 ng/mL). Bars with ** denotes statistically different at the P < 0.01 level. | |
{kind=link}
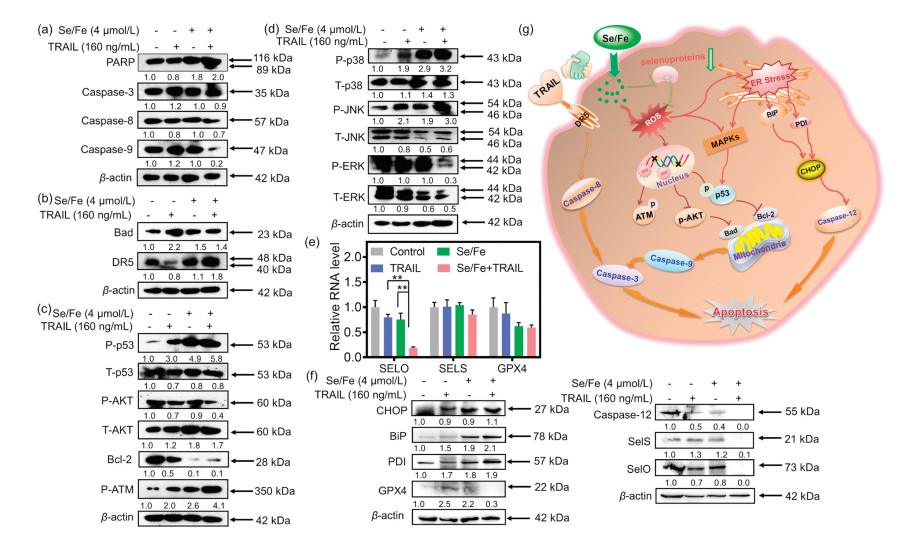
Substantial evidences have delineated that ROS could induce caspase-dependent apoptosis through receptor-mediated extrinsic signaling pathways or mitochondria-mediated intrinsic signaling pathways [44, 45]. As shown in Figs. 6a and b, compared to single treatment, the combination treatment could more effectively trigger the cleavage of PARP, activate caspase-3, -8, -9 and up-regulate the expression of death receptor 5 (DR5), which shows both exogenous and endogenous apoptotic pathways lead to cell apoptosis. Meanwhile, since modulation of ROS/MAPK signaling pathways could also lead to caspase-dependent cell apoptosis [46, 47], we explored related signaling pathways including p53, anti-apoptotic kinases AKT and mitogen-activated protein kinase (MAPKs) pathways. It was found that the combination treatment could induce the up-regulation of phosphorylated p53 (P-p53) and inactivate anti-apoptotic kinases AKT (P-AKT down-regulation). It is known that P-p53 and the inactivation of AKT regulate the expressions of Bcl-2 family members composed of anti-apoptosis proteins (Bcl-2 and Bcl-xL), and pro-apoptotic proteins (Bax, Bad and Bid) [48, 49]. As displayed in Figs. 6b and c, the combination treatment remarkably induced the overexpression of pro-apoptosis Bcl-2 family protein Bad and down-regulated the pro-survival Bcl-2 family proteins Bcl-2. Meanwhile, the combination treatment upregulated the expression of phosphorylation ATM (P-ATM), which is closely relevant to DNA damage, induced the accumulation of ROS [50]. What is more, as one of the complex intracellular signaling pathways, MAPK cascade pathway including p38, JNK, ERK, plays critical roles in the action of anticancer agents by converging extracellular signals and causing cell apoptosis [51]. Our evidence shows that the expression levels of p38, JNK, ERK have changed in cancer cells with the co-treatment (Fig. 6d). The co-treatment leads to a remarkable increase on phosphorylation of the proapoptotic kinases p38 and JNK. By contrast, the co-treatment brought about the inactivation of anti-apoptotic kinases ERK. These results confirm that MAPKs signaling pathways play an essential role in the execution of apoptosis through regulating ROS generation caused by the co-treatment. Altogether, our developed Se/Fe complex facilitates TRAIL treatment mainly through inducing the robust intracellular generation of ROS to regulate the expressions of multiple molecules related to extrinsic and intrinsic pathways for sensitizing tumor cells.

|
Download:
|
| Fig. 6. Se/Fe complex facilitates TRAIL treatment against HeLa cells through triggering extrinsic- and intrinsic-dependent apoptosis signaling pathways and inducing ER stress. (a) The effects of Se/Fe complex and/or TRAIL on the activation of PARP and Caspase 3/8/9. (b) The effects of Se/Fe complex and/or TRAIL on the expression levels of DR5 and Bad. (c) The effects of Se/Fe complex and/or TRAIL on the expression levels of p53, AKT, Bcl-2and P-ATM were detected by western blot analysis. (d) The change of Se/Fe complex and/or TRAIL on the phosphorylation status and the expression level of MAPKs. (e) The synergistical effect of Se/Fe complex and TRAIL challenge on the relative RNA levels of selenoprotein genes (SELO, SELS, GPX4) in HeLa cells treated with Se/Fe complex or/and TRAIL (160 ng/mL) for 12 h. ** represent statistically different at the P < 0.01 level respectively. (f) The expression levels analysis of selenoproteins and the ER Stress-related proteins by the effects of Se/Fe complex or/and TRAIL (160 ng/mL) in HeLa cells. Cells were treated with 4 μmol/L Se/Fe complex for 24 h, followed by 160 ng/mL TRAIL for 24 h. (g) Proposed schematic diagram of the underlying mechanism of Se/Fe complex combined TRAIL treatment. Cells were pre-treated with 4 μmol/L Se/Fe complex for 24 h, followed by the treatment of TRAIL (160 ng/mL) for another 24 h. | |
{kind=link}
As above evidences indicated that Se/Fe complex with good penetration capability could effectively inhibit the viability of HeLa cells and its migration/invasion capabilities through downregulating ER-stress related selenoproteins, we subsequently investigated the effects of the combination treatment on the expressions of related selenoproteins to uncover its underlying mechanism. The quantitative PCR analysis demonstrated that combination treatment could more efficiently reduce the expressions of multiple selenoproteins involving SELO, SELS, GPX4 especially SELO (decreased by 76.5%) compared to the complex or TRAIL treatment alone (Fig. 6e). Actually, evidences have demonstrated that some selenoproteins such as GPX4 and SELO could regulate cellular redox balance and protect cells from oxidative damage [35, 36, 52]. In addition, it was reported that SELS suppression could enhance ER stress-induced cell apoptosis [33, 53].
After that, we carefully evaluated the changes of ER stress post treatment using western blotting assay. It was found that the expression of the CCAAT/enhancer binding protein homologous protein (CHOP, one of the typical markers of ER stress) was significantly up-regulated (Fig. 6f). Meanwhile, the ER chaperone protein binding immunoglobulin protein (BiP) and protein disulfide isomerase (PDI) proteins, which playing critical roles in protein synthesis and misfolded proteins reconstruction, were upregulated and would promote the proapoptotic functions of ER stress pathway after treatment (Fig. 6f). Furthermore, the enhanced cleavage of pro-caspase-12 also confirmed the cell apoptosis induced by ER stress. In general, as illustrated in Fig. 6g, Se/Fe complex synergistically stimulates TRAIL-induced apoptosis by suppressing the expression of selenoproteins, which also triggers endoplasmic reticulum stress and activates mitochondrial pathway.
In summary, we have developed a safe and dual-functional Se/ Fe complex for TRAIL-based cancer treatment. The Se/Fe complex with low toxicity to normal cells and good penetration capability could serve as anticancer agent to significantly inhibit HeLa cells proliferation and invasion capabilities through down-regulating the ER stress related selenoproteins and destroy tumor spheroids. In addition, it could greatly enhance the sensitization of HeLa cells to TRAIL-based treatment. Distinguished from TRAIL treatment alone which exhibiting poor inhibition efficacy, the complex synergizing with TRAIL treatment could more efficiently kill tumor cells via inducing the robust generation of intracellular ROS, down-regulating ER stress related selenoproteins to promote ER stress for triggering tumor cells apoptosis in extrinsic and intrinsic signaling pathways. This study provides a safe and effective chemo-drug and sensitizer for optimizing TRAIL-based cancer treatment.
Declaration of competing interestThe authors declare that they have no conflict of interest.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21877049, 31871006), National Program for Support of Top-notch Young Professionals (No. W02070191), Major Program for Tackling Key Problems of Industrial Technology in Guangzhou (No. 201902020013), Dedicated Fund for Promoting High-Quality Marine Economic Development in Guangdong Province (No. GDOE-2019-A31).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.03.004.
| [1] |
S. von Karstedt, A. Montinaro, H. Walczak, Nat. Rev. Cancer 17 (2017) 352-366. DOI:10.1038/nrc.2017.28 |
| [2] |
G.L. Wang, X.M. Wang, H. Yu, et al., Nat. Chem. Biol. 9 (2013) 84-89. DOI:10.1038/nchembio.1153 |
| [3] |
J. Wiezorek, P. Holland, J. Graves, Clin. Cancer Res. 16 (2010) 1701-1708. DOI:10.1158/1078-0432.CCR-09-1692 |
| [4] |
J. Eberle, Cancers 11 (2019) 656. DOI:10.3390/cancers11050656 |
| [5] |
M.L. Plissonnier, S. Fauconnet, H. Bittard, et al., Oncotarget 8 (2017) 107744-107762. DOI:10.18632/oncotarget.22632 |
| [6] |
S.R. Im, Y.J. Jang, Biochem. Biophys. Res. Commun. 424 (2012) 65-70. DOI:10.1016/j.bbrc.2012.06.067 |
| [7] |
S.C. Gupta, S. Reuter, K. Phromnoi, et al., J. Biol. Chem. 286 (2011) 1134-1146. DOI:10.1074/jbc.M110.191379 |
| [8] |
X.F. Chen, H. Thakkar, F. Tyan, et al., Oncogene 20 (2001) 6073-6083. DOI:10.1038/sj.onc.1204736 |
| [9] |
K. Selvarajoo, Prog. Biophys. Mol. Biol. 128 (2017) 142-154. DOI:10.1016/j.pbiomolbio.2017.02.009 |
| [10] |
X. Li, M. You, Y.J. Liu, et al., Sci. Rep. 7 (2017) 42748. DOI:10.1038/srep42748 |
| [11] |
C.J. Liang, Y.C. Xu, G.M. Li, et al., OncoTargets Ther. 10 (2017) 417-428. DOI:10.2147/OTT.S127202 |
| [12] |
J.R. Cubillos-Ruiz, S.E. Bettigole, L.H. Glimcher, Cell 168 (2017) 692-706. DOI:10.1016/j.cell.2016.12.004 |
| [13] |
R. Martin-Perez, M. Niwa, A. Lopez-Rivas, Apoptosis 17 (2012) 349-363. DOI:10.1007/s10495-011-0673-2 |
| [14] |
R. Iurlaro, C. Munoz-Pinedo, FEBS J. 283 (2016) 2640-2652. DOI:10.1111/febs.13598 |
| [15] |
C. Munoz-Pinedo, A. Lopez-Rivas, Cell Death Differ. 25 (2018) 226. DOI:10.1038/cdd.2017.155 |
| [16] |
F. Dufour, T. Rattier, A.A. Constantinescu, et al., Oncotarget 8 (2017) 9974-9985. DOI:10.18632/oncotarget.14285 |
| [17] |
Z.Q. Deng, L.L. Yu, W.Q. Cao, et al., Chem. Commun. 51 (2015) 2637-2640. DOI:10.1039/C4CC07926D |
| [18] |
H.Q. Lai, X. Zhang, P.J. Feng, et al., Chem. Asian J. 12 (2017) 982-987. DOI:10.1002/asia.201700272 |
| [19] |
D.L. Zeng, S.L. Deng, C.C. Sang, et al., Bioconjugate Chem. 29 (2018) 2039-2049. DOI:10.1021/acs.bioconjchem.8b00247 |
| [20] |
E.S.J. Arner, Free Radical Biol. Med. 120 (2018) S31. |
| [21] |
J.C. Avery, P.R. Hoffmann, Nutrients 10 (2018) 1203. DOI:10.3390/nu10091203 |
| [22] |
Y.L. Ye, F. Fu, X.M. Li, et al., J. Cell. Biochem. 117 (2016) 106-117. DOI:10.1002/jcb.25254 |
| [23] |
H.Y. Huang, B.L. Yu, P.Y. Zhang, et al., Angew. Chem. Int. Ed. 54 (2015) 14049-14052. DOI:10.1002/anie.201507800 |
| [24] |
Q. Chen, L.Z. Feng, J.J. Liu, et al., Adv. Mater. 28 (2016) 7129-7136. DOI:10.1002/adma.201601902 |
| [25] |
N. Yamamoto, A.K. Renfrew, B.J. Kim, et al., J. Med. Chem. 55 (2012) 11013-11021. DOI:10.1021/jm3014713 |
| [26] |
W. Tao, X.Y. Ji, X.B. Zhu, et al., Adv. Mater. 30 (2018) e1802061. DOI:10.1002/adma.201802061 |
| [27] |
J.J. Chen, Z.D. Luo, Z.N. Zhao, et al., Biomaterials 71 (2015) 168-177. DOI:10.1016/j.biomaterials.2015.08.031 |
| [28] |
X. Fu, Y. Yang, X. Li, et al., Nanomedicine 12 (2016) 1627-1639. DOI:10.1016/j.nano.2016.01.012 |
| [29] |
W. Cao, W. Zheng, T. Chen, Sci. Rep. 5 (2015) 1-11. |
| [30] |
M.P. Rayman, Lancet 379 (2012) 1256-1268. DOI:10.1016/S0140-6736(11)61452-9 |
| [31] |
G.F. Combs, Nutrients 7 (2015) 2209-2236. DOI:10.3390/nu7042209 |
| [32] |
H. Liu, H. Xu, K. Huang, Metallomics 9 (2017) 21-37. DOI:10.1039/C6MT00195E |
| [33] |
S.Q. Du, H.M. Liu, K.X. Huang, Biochim. Biophys. Acta:Gen. Subj. 1800 (2010) 511-517. DOI:10.1016/j.bbagen.2010.01.005 |
| [34] |
S. Arbogast, A. Ferreiro, Antioxid. Redox Signaling 12 (2010) 893-904. DOI:10.1089/ars.2009.2890 |
| [35] |
H. Steinbrenner, B. Speckmann, L.O. Klotz, Arch. Biochem. Biophys. 595 (2016) 113-119. DOI:10.1016/j.abb.2015.06.024 |
| [36] |
S. Florian, S. Krehl, M. Loewinger, et al., Free Radical Biol. Med. 49 (2010) 1694-1702. DOI:10.1016/j.freeradbiomed.2010.08.029 |
| [37] |
S.J. Han, B.C. Lee, S.H. Yim, et al., PLoS One 9 (2014) e95518. DOI:10.1371/journal.pone.0095518 |
| [38] |
C. Hetz, Nat. Rev. Mol. Cell Biol. 13 (2012) 89-102. DOI:10.1038/nrm3270 |
| [39] |
H. Pelicano, D. Carney, P. Huang, Drug Resist. Updat. 7 (2004) 97-110. DOI:10.1016/j.drup.2004.01.004 |
| [40] |
W. Tao, N. Kong, X. Ji, et al., Chem. Soc. Rev. 48 (2019) 2891-2912. DOI:10.1039/C8CS00823J |
| [41] |
X.Y. Ji, Y. Kang, J. Ouyang, et al., Adv. Sci. 6 (2019) 1901211. DOI:10.1002/advs.201901211 |
| [42] |
H.X. Liu, W.Q. Lin, L.Z. He, T.F. Chen, Biomaterials 226 (2020) 119545. DOI:10.1016/j.biomaterials.2019.119545 |
| [43] |
Y. Ren, Q.S. Sun, Z.G. Yuan, Y.Y. Jiang, Chin. Chem. Lett. 30 (2019) 1233-1236. DOI:10.1016/j.cclet.2019.03.029 |
| [44] |
M.L. Circu, T.Y. Aw, Free Radical Biol. Med. 48 (2010) 749-762. DOI:10.1016/j.freeradbiomed.2009.12.022 |
| [45] |
M. Redza-Dutordoir, D.A. Averill-Bates, Biochim. Biophys. Acta:Mol. Cell Res. 1863 (2016) 2977-2992. DOI:10.1016/j.bbamcr.2016.09.012 |
| [46] |
L.Z. He, S.B. Ji, H.Q. Lai, T.F. Chen, J. Mater, Chem. B 3 (2015) 8383-8393. |
| [47] |
Y.H. Yang, Q. Xie, Z.N. Zhao, et al., ACS Appl. Mater. Interfaces 9 (2017) 25857-25869. DOI:10.1021/acsami.7b07167 |
| [48] |
T. Chen, Y.S. Wong, Int. J. Biochem. Cell Biol. 41 (2009) 666-676. DOI:10.1016/j.biocel.2008.07.014 |
| [49] |
B.S. Tan, K.H. Tiong, H.L. Choo, et al., Cell Death Dis. 6 (2015) e1826. DOI:10.1038/cddis.2015.191 |
| [50] |
A.N. Blackford, S.P. Jackson, Mol. Cell 66 (2017) 801-817. DOI:10.1016/j.molcel.2017.05.015 |
| [51] |
N. Wang, Y.X. Feng, L.L. Zeng, et al., ACS Appl. Mater. Interfaces 7 (2015) 14933-14945. DOI:10.1021/acsami.5b03739 |
| [52] |
M. Roman, P. Jitaru, C. Barbante, Metallomics 6 (2014) 25-54. DOI:10.1039/C3MT00185G |
| [53] |
Y. Gao, H.C. Feng, K. Walder, et al., FEBS Lett. 563 (2004) 185-190. DOI:10.1016/S0014-5793(04)00296-0 |