 2020, Vol. 31
2020, Vol. 31 


b School of Pharmacy, Ji'nan University, Guangzhou 510632, China;
c Department of Medicinal Chemistry, Key Laboratory of Chemical Biology(Ministry of Education), School of Pharmaceutical Sciences, Shandong University, Ji'nan 250012, China;
d Department of Chemistry, Jiangsu Key Laboratory of Drug Design and Optimization, China Pharmaceutical University, Nanjing 211198, China;
e State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, and Collaborative Innovation Center of Biotherapy, Sichuan University, Chengdu 610041, China;
f State Key Laboratory of Natural Medicines, Jiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, China Pharmaceutical University, Nanjing 210009, China;
g Key Laboratory of Drug Targeting and Drug Delivery System of Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, China;
h Key Laboratory of Pesticide & Chemical Biology(CCNU), Ministry of Education, Department of Chemistry, Central China Normal University, Wuhan 430079, China
Li Rao*
Protein ligand refers to any molecule that binds to a protein, like substrates, cofactors, metal ions or inhibitors. Many of the protein's functions originate from their binding toward specific ligand molecules, like enzymatic catalysis begins with binding of substrates. It is worthy of mention that the so called targeted drugs rely on selective binding toward the targeted proteins. Thus a deep understanding of protein-ligand interaction would be of great help to drug design purpose.
The two major topics in protein-ligand interaction study are binding structure and binding affinity. Protein-ligand binding structure is the structural basis for the exploration of proteinligand interaction. For the moment, accurate determination of protein-ligand complex 3D structure severely relies on X-ray crystallographic experiments, which are expensive and could be extremely difficult in some cases. Thus binding structure theoretical prediction becomes an appealing alternative, like the widely used molecular docking. Theoretical calculation of proteinligand binding structure includes two major steps, with the first one searching the potential energy surface of protein-ligand complex for all of (or most of) the possible binding conformations, followed by the second step in which all conformations are ranked to reveal the optimal binding structure. Molecular docking methods search binding conformations on a molecular mechanics level of theory, usually employing semi-flexible approximation for the simplification of conformation space. Then the optimal binding structure is revealed according to ranking of binding affinity estimated using an empirical scoring function. As one of the most popular CADD tool, molecular docking is known for its efficiency and guarantee to produce a result, unlike the uncertainty of crystallographic experiment, although the reliability of the results are questionable, unlike crystallographic experiment again. Plewczynski et al. reported a systematic assessment [1] of seven main stream molecular docking methods (eHiTS, FlexX, Glide, GOLD, LigandFit, Surflex, AutoDock) [2-9]. Employing a success standard of RMSD < 2 Å, they found out that the docking success rate lies between 40%–60% for small ligand molecules and decreased to 20%–50% for large ligand molecules. Note that the ligands could be positioned at the right place but interact with wrong residue in the predicted binding structure with RMSD < 2 Å.
On the other hand, Plewczynski et al. also found that molecular docking tools were able to produce at least one good pose (RMSD < 2 Å) in 80% of the calculation, but failed to recognize its importance owing to the deficiency of empirical scoring function. Therefore replacing empirical scoring function with more accurate theory for binding conformation ranking should be able to increase docking accuracy. Recently, our group developed a protocol named DOX for accurate protein-ligand binding structure prediction [10, 11], in which first principle method was used to rank the binding poses instead of empirical scoring function. The DOX protocol is a sophisticated combination of molecular docking, semi-empirical quantum chemistry and first principle density functional theory, based on CSAMP (conformation search across multiple-level potential-energy surfaces) strategy. CSAMP strategy refers to joint application of coarse level conformation search, medium level conformation optimization/screen, and fine level conformation ranking (Fig. 1). Such strategy elevates ranking accuracy to fine level, while avoiding search the entire potential energy surface with expensive file level theory. The first version of DOX protocol was announced in 2016 [10]. The second version was reported recently, together with a systematic validation [11]. In the latest version of DOX protocol, Surflex docking is used to generate a total of 300 diverse binding poses, covering most of (if not all) the conformation space. In the next step, semi-empirical theory PM7 is used to optimize each binding pose and screen out the majority of unlikely poses. The left top 10 poses are re-optimized and credited employing extended ONIOM (XO) method to reveal the top 1 pose. XO method is a hybrid chemistry theory developed by Xu et al., which enables first principle description of protein-ligand complex via divide & conquer strategy [12].

|
Download:
|
| Fig. 1. A schematic illustration of CSAMP strategy and DOX protocol. | |
{kind=link}
The latest version of DOX protocol was validated against the Astex test set including 85 protein-ligand complex crystal structures. Employing RMSD < 2 Å as success standard, DOX prediction achieved an impressive success rate of 99%, indicating a significant improvement over traditional molecular docking method. It is worthy of mention that employing a harsh standard of RMSD < 1 Å, DOX prediction still achieved a success rate of 70%, suggesting that the reliability of DOX protocol is close to costly crystallographic experiment. Owing to the employment of first principle calculations, DOX protocol is much more computational demanding than traditional docking. Thus it could not replace docking in large scale virtual screen. In scenarios like enzymatic mechanism study or lead structure optimization, DOX would be a cost-efficient option. For tryout of the DOX protocol, a web server can be accessed at http://202.114.32.71:10280/wandox/DOXserver/home.html.
The work of this section was supported by the National Natural Science Foundation of China (Nos. 21603081, 21873035).
2. Virtual target profiling in lead discovery and drug repositioningGuobo Li*
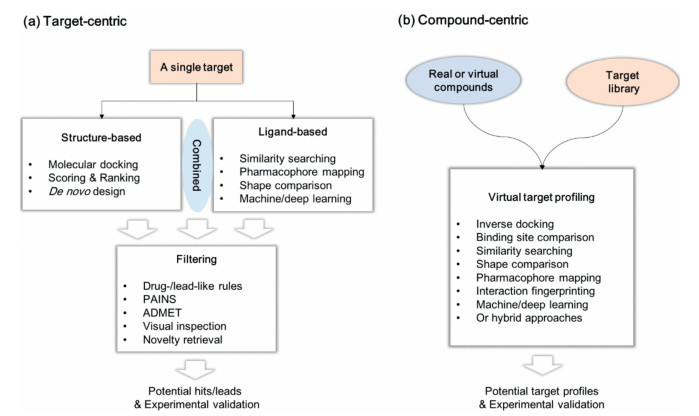
Computer-aided drug design (CADD) tools have been involved in target-centric and compound-centric drug discovery pipelines (Fig. 2), which considerably improve the efficiency of drug discovery process and even impact evolution of the birth of new drugs. The target-centric manner usually begins with defining a model to represent a drug target, followed by screening or creating compounds to fit the model, and identifying potential hits/leads for experimental validation. In this paradigm, various structure- and ligand-based approaches have been well-established and wideused, such as molecular docking, scoring/re-scoring, de novo design, similarity searching, shape recognition, pharmacophore mapping, machine/deep learning or any combination thereof (Fig. 2A) [13-15]. The effectiveness of these approaches has been continuously demonstrated [16-18]. Relatively, a compoundcentric strategy (Fig. 2B) is emerging and gradually becoming popular, for example, virtual target profiling (also termed as target prediction, in silico target identification or in silico target fishing), which enables a parallel implementation of interrogating molecules of interest (MOIs) against various targets in a single virtual screening campaign. Virtual target profiling is not only useful for discovering high-quality hits/leads but also helpful for providing useful information to identify new targets for an existing drug and thereby exploit additional medical applications [19, 20].

|
Download:
|
| Fig. 2. Target- and compound-centric strategies. (a) Commonly used target-centric drug design methods; (b) Emerging compound-centric virtual targeting profiling strategies. | |
{kind=link}
To date, there have been a number of sophisticated virtual target profiling tools. Almost two decades ago, Chen et al. proposed an inverse molecular docking approach termed INVDOCK [21], which could automatically dock MOIs to different target binding sites in the target library and prioritize all retrieved targets using a scoring function. Afterwards, several other inverse molecular docking tools have been reported, such as TarFisDock [22], idTarget [23], and iRAISE [24]. These methods have been successfully used in drug repositioning and hit/lead discovery. Pharmacophore, representing the spatial arrangement of features essential for a molecule to interact with a specific target receptor, was used as an alternative method for virtual target profiling. PharmMapper is the most representative tool, and a user-friendly, freely accessed webserver is available [25, 26]. Besides, on basis of the assumption that structurally similar proteins or binding sites may be bound with similar ligands, a variety of binding site comparison methods have been established, such as SiteBase [27] and SiteAlign [28], which may be possibly coupled with pocket analysis methods such as D3Pockets [29] to provide additional information for virtual target profiling.
Apart from above methods, ligand chemical similarity has been commonly used for predicting potential targets for MOIs by comparing their chemical structures with a prepared database of compounds whose targets are known. For example, the similarity ensemble approach (SEA) proposed by Keiser et al. works through quantitatively grouping related protein targets on basis of the similarity of 2D fingerprints decoding their ligand chemical structures [30, 31]. Application of SEA led to identification of several new drug-target associations [31], largely demonstrating its effectiveness. In recent years, a number of 3D similarity methods have been also established for virtual target profiling, such as ChemMapper [32] and ReverseScreen3D [33], further expanding the usefulness and scope of ligand chemical similarity in virtual target profiling.
Each virtual target profiling method has its own features and advantages, but may still suffer some limitations. Combination of different, in particular complementary methods, has potential to improve the predictive ability. For instance, inverse molecular docking tools usually use a 'universal' scoring function to predict binding affinity and prioritize all retrieved targets from a prepared target library for the MOIs; sometimes, many of these tools fail to correctly differentiate true binding proteins for the MOIs from a large set of retrieved targets, since they probably do not specifically consider the key features essential for binding with each target. We previously proposed a combined molecular docking and pharmacophore modeling (i.e., partly represent the target-specific binding features) for virtual target profiling, which showed improved predictive ability [34]. Recently, we developed a target-customized virtual target profiling method termed IFPTarget [35]. It employs a protein-ligand interaction fingerprinting method to analyze the target-specific binding features, and also integrates a machine learning-based scoring method for binding affinity prediction [35, 36]. When applied to screen against our established target library, IFPTarget showed good prediction ability to retrieve known targets within the top-ranked list, which was further examined by wet experiments [35, 37].
Family-focused virtual target profiling will be an entirely worthwhile direction. Recently, Jiang and Zheng's group developed a kinome-wide virtual target profiling platform, termed KinomeX, which is made by a multi-task deep neural network model established based on 140, 000 bioactivity data points for 391 kinases [38]. We believe that the constant development of new virtual target profiling methods, coupled with advances/achievements in biochemistry, medicinal chemistry, structural biology and many other fields (e.g., artificial intelligence), will improve the efficiency of high-quality lead/drug discovery and drug repositioning.
The author thanks the financial support from the National Natural Science Foundation of China (No. 81874291), Outstanding Interdiscipline Project of West China Hospital of Sichuan University (No. ZYJC18024) and the Fundamental Research Funds for the Central Universities.
3. Introduction on in silico autophagy methods and strategies in medicinal chemistryGuan Wang, Liang Ouyang*
Autophagy is a conserved cellular physiological catabolic process in eukaryotes responsible for degradation and recycling of damaged organelles and long-lived proteins [39]. Over the past decades, accumulated experimental evidence has indicated that autophagy can be considered as a potential target for medicinal research, and regulation of autophagy has a certain therapeutic effect in various diseases and model organisms [40]. So far, there have been about 40, 000 researches on the field of autophagy. However, there are still some autophagy processes, mechanisms or targets that cannot be analyzed by experiments. Hence, in recent years many articles using web servers, mathematical models, and systems biology methods for autophagy research have been published, which were known as the "in silico autophagy method". This editorial focuses on these new methods and strategies in autophagy research to promote the use of in silico methods in medicinal research, especially, autophagy-related drug target discovery and drug design.
In recent years, the emergence of many new web servers and database has provided us with convenient use and comprehensive information in autophagy research. Wang et al. established a docking preferred-protein selection server protein structure selection (ProSelection) to accelerate the selection of proper protein structure [41]. Autophagic compound target prediction (ACTP) was designed by Xie et al. to predict potential autophagyrelated targets and pathways of small-molecule compounds and provide reverse docking [42]. Some statistical databases were also helpful for researchers to understand autophagy-related proteins and compounds. Jacomin et al. provided a web server iLIR to identify the microtubule-associated protein 1 light chain 3- interacting region-containing proteins (LIRCPs) in 8 organisms [43]. Deng et al. developed the autophagic compound database (ACDB) with targets, pathways and diseases information of 357 autophagic compounds for researchers to refer to [44]. Both Human Autophagy Modulator Database (HAMdb) and Autophagy Small Molecule Database (AutophagySMDB) have collected a large number of autophagic proteins and compounds [45, 46].
Due to the limitations of current experimental techniques and methods, many mathematical models have been applied to study autophagy mechanism and related disease. Börlin et al. reported an agent-based model (ABM) could measure short-term autophagic flux and cell-to-cell variability, and revealed the dynamics of spatio-temporal autophagy [47]. Tavassoly et al. built a dynamic model to study the interaction between apoptosis and autophagy successfully in mammalian cells [48]. The Petri net model of xenophagy was presented by Scheidel et al. to clarify the key defense mechanism of Salmonilla, provided a guide for study of Salmonella xenophagy pathway [49]. Aslan et al. demonstrated the conservation of autophagy-related proteins in ciliates through the integration analysis of several databases and resources [50]. Awan et al. established an extensive in silico method to predict autophagy-related proteins deleterious missense mutation on hepatocellular carcinoma [51]. A multilevel hybrid-modeling paradigm was designed to simulate the mitochondrial dynamics and normal aging physiological parameters in degenerative aging [52].
Complex disease such as cardiovascular disease and cancer is difficult to treat effectively with a single target. And highly specific drugs may produce unexpected side effects or exhibit lower therapeutic effects. However, the development of systems biology has brought dawn to researchers. Zhou et al. used gene microarray of tissue samples from patients to find that autophagy-related gene 7 variant rs8154 could be identified as a prognostic indicator for breast cancer [53]. Similarly, a global quantitative phosphoproteomic approach was performed to verify a novel function of TBK1 induced mitophagy through PINK-Parkin pathway [54]. In the study by Xuan et al., a cacybp-related protein interaction network was constructed using the human glioma database, and 121 differentially expressed genes were identified. It was found that Cacybp expression in low-grade glioma and glioblastoma was no significant, which could not be the prognosis marker [55]. In the comprehensive review by Lai group, new methods to identify drug targets by systems biology were discussed in detail [56].
In addition to the above-mentioned methods specifically applied in the autophagy research, there are also many new technologies and strategies of drug design that can greatly promote research progress in this field. Lu et al. have provided an overview of computational and experimental approaches involved in allosteric sites identification of proteins [57]. Yang et al. introduced the artificial intelligence (AI) methods, summarized machine learning and deep learning approaches in drug discovery and development scenarios [58].
Currently, due to the development of computer technology, omics techniques, the accumulation of medicinal chemistry, pharmacology, and pathology data, in silico autophagy methods will play an increasingly important role in the medicinal research. Particularly, advanced techniques and strategies such as AI, especially deep learning algorithms, can effectively help us to continuously explore human diseases from aspects of protein binding as well as the molecular mechanism, will further boost the application of in silico methods in autophagy-related mechanisms and interventions. Although in silico approaches are still in their infancy, there is no doubt that increasing advanced in silico methods will be developed and combined with experiments to solve the problem of autophagy research. Looking further afield, we look forward to witnessing innovative, revolutionary and subversive achievements in the area of autophagy and its application in drug discovery.
4. Defatty-acylase activity of histone deacetylases: New directions for drug designJintong Dua, Xuben Houb, *, Hao Fangb, *
Reversible acylation of lysine, which has been recognized as an important post-translational modification (PTM), has emerged as a basilic regulatory mechanism in mammalian. Acylation state of proteins is modulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs) [59]. Mammalian HDACs can be divided into four phylogenetic classes based on sequence similarity and enzymatic mechanism. The zinc-dependent hydrolases are categorized into class I (HDAC1, 2, 3, and 8), IIa (HDAC4, 5, 7 and 9), IIba (HDAC6 and 10) and IV (HDAC11). Class Ⅲ HDACs (also known as Sirtuins) are a distinct group of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases (SIRT1-7) [60].
The initially discovered biological functions of HDACs mainly achieved through regulation of acetyllysine levels in histones and non-histone proteins. Recently, it has become evident that ε-N atom of lysine sidechain could be modified with various acyl groups other than acetyl [61]. Specially, the long-chain lysine ε-Nacylation (fatty-acylation) plays an important role in multiple cellular processes including membrane targeting, cell signaling and protein-protein interactions [62]. Dysregulation of protein fatty-acylation is implicated in disease such as cancer, neurodegenerative diseases and cardiovascular disease [63, 64]. Interestingly, recent studies demonstrate that certain HDACs can remove fatty acylation and serve as proficient fatty acid deacylases [61]. Specially, these newly revealed defatty-acylation activities of HDACs provide new directions for the design of specific modulators.
Here, we reviewed the recent advances in deacylase activities of HDACs as well as the structural basis for the binding of acylated substrates. Four acyl groups with different hydrocarbon lengths, including acetyl (C2), hexanoyl (C5), decanoyl (C9) and myristoyl (C13), will be discussed in this paper. Moreover, we also present examples for the design of isoform selective HDAC inhibitors based on the defatty-acylation activity.
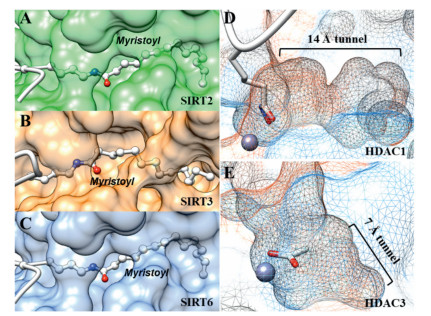
The deacylase activity was first discovered in NAD+-dependeted class Ⅲ HDACs (SIRT1-7) [61]. Using a panel of acylated lysine substrate, John M. Denu's group and Christian A. Olsen's group revealed that defatty-acylase activity seems to be an intrinsic enzymatic activity of mammalian Sirtuins [65-67] (Fig. 3). SIRT1-3 displayed strong deacylation activity against acetyl substrate as well as long-chain acyl groups. Other four Sirtuins (SIRT4-7) have very weak deacetylase activity in vitro. Interestingly, SIRT4 and SIRT6 preferentially hydrolyzes long-chain fatty acyl groups (e.g., hexanoyl, decanoyl and myristoyl), relative to acetyl group [68, 69]. SIRT5 preferentially hydrolyses decanoyl lysine than hexanoyl and myristoyl lysine. In particular, deacetylation activity of SIRT6 could be activated by free long-chain fatty acids [65]. The deacylase activity of SIRT7 cannot be detected in vitro, though its biological functions have been attributed to deacylase activity. Hening Lin's group demonstrate that double-stranded DNA (dsDNA) can significantly improve deacetylase activity of SIRT7 [70]. Recently, they further revealed that RNA is potent SIRT7 activator in vitro against fatty acyl groups [71] (Fig. 3). Crystal structures of SIRT1-3 already revealed the existence of extended binding pockets for the binding of fatty acid acylated lysine substrates (Figs. 4A–C).

|
Download:
|
| Fig. 3. Heatmap showing the activities of HDAC1-11 and SIRT1-7 against different acylated substrates. Data from literatures [65, 66, 72, 73, 76]. The deacylase activity of SIRT7 is detected in the presence of RNA [71]. | |
{kind=link}

|
Download:
|
| Fig. 4. Crystal structures of HDACs revealed the structure basis for the binding of fatty-actylated substrate. (A) SIRT2 complexed with myristoylated-lysine substrate (PDB 4Y6L). (B) SIRT3 complexed with myristoylated-lysine substrate (PDB 5BWN). (C) SIRT6 complexed with myristoylated-lysine substrate (PDB 3ZG6). (D) Internal cavity of HDAC1 (PDB 5ICN). (E) Internal cavity of HDAC3 (PDB 4A69). | |
{kind=link}
Inspired by these finding for the Sirtuins, researchers envisioned that the ability to cleave long-chain acyl modifications might also be the case for the zinc-dependent HDACs (HDAC1-11). As a class I HDAC member, HDAC8 exhibit low deacetylation activity in vitro compared to other HDACs. Hening Lin's group first observed increased catalytic efficiency of HDAC8 towards fatty acyl groups, due to the lower Km values [72]. Christian A. Olsen's group screened the activity of all zinc-dependent HDACs (HDAC1-11) against a series of ε-N-acyllysine substrates and observed different deacylase activities [67, 73]. As shown in Fig. 3, HDAC1-3 possess robust deacetylation activity, however, the catalytic efficiencies of HDAC1-3 decrease as the length of acyl group increase (Fig. 3). The crystal structure of HDAC1 revealed the existence of a 14 Å internal cavity in the bottom of lysine binding site [74, 75], which could accommodate the binding of fatty-acid acylated substrates (Fig. 4D). A similar big internal cavity could also be observed in crystal structure of HDAC2 [75]. Interestingly, HDAC3 also possess an internal cavity, however, the volume is smaller than those in HDAC1 and HDAC2 (Fig. 4E). This structural difference may explain the reason why HDAC3 showed weaker catalytic efficiency toward myristoyl (C13) lysine than hexanoyl (C5) lysine. The class IIa isozymes (HDAC4, 5, 7, and 9) exhibited no activities against acyllysine substrates in vitro. The class IIb enzyme (HDAC6 and HDAC10) showed moderate deacetylase activity and did not show efficiency defatty-acylase activity. Interestingly, HDAC11, the only member of class IV HDAC, exhibit robust activity against longchain acyl groups [76].
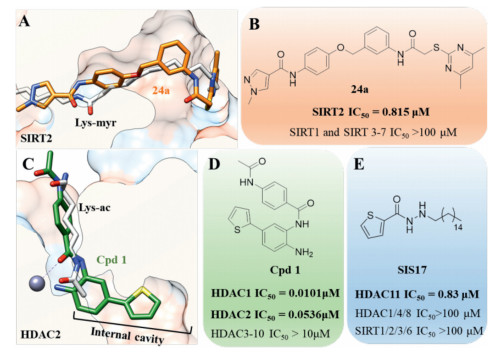
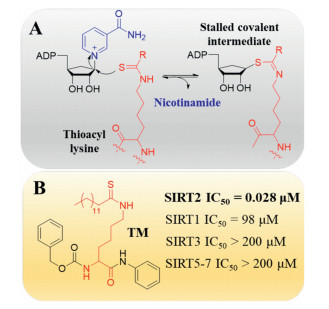
Most known Sirtuin inhibitors lack of enough potency as well as isoform selectivity. Therefore, developing more potent and selective inhibitors would greatly benefit the investigation of therapeutic potential of Sirtuins. Recently, Yang et al. identified a series of N-(3-(phenoxymethyl)phenyl)acetamide derivatives as selective SIRT2 inhibitors [77, 78]. Crystal structure indicated that the most potent and selective inhibitor 24a bind to an enlarged hydrophobic pocket in SIRT2. Particularly, the binding mode of 24a mimics that of myristoylated-lysine substrates (Figs. 5A and B) [78], which may account for its good isoform selectivity. Thioacyl lysine analogs can react with NAD+ and form a relatively stable intermediate, therefore provide a valuable strategy for covalent inhibition of Sirtuins (Fig. 6A) [79]. According to this mechanism, a series of fatty-acid substrate mimetics, including thiomyristoyl lysine (TM) [80] and its derivatives [81, 82], were recently developed as mechanism-based potent and selective SIRT2 inhibitors (Fig. 6B).

|
Download:
|
| Fig. 5. Isoform selective HDAC inhibitors based on defatty-acylation activity. (A) Comparison of the binding mode of myristoyl lysine (PDB 4Y6L) and 24a (PDB 5YQO) in SIRT2. (B) Structure and inhibitory activity of 24a. (C) Comparison of the binding mode of acetyl lysine and Cpd1 (PDB 4LY1) in HDAC2. The binding mode of acetyl lysine was derived from HDAC8 crystal structure (PDB 2V5W). Structures and inhibitory activities of Cpd 1 (D) and SIS17 (E). μM: μmol/L. | |
{kind=link}

|
Download:
|
| Fig. 6. (A) Mechanism-based inhibition of Sirtuins by thioacyl lysine analogs. (B) Structure and activity of SIRT2 selective inhibitor TM. μM: μmol/L. | |
{kind=link}
Although the crystal structure of zinc-dependent HDACs with fatty-acylated substrate has not been determined, the internal cavity in class I HDACs (Figs. 4D and 5C), which has been previously recognized as the acetic acid releasing channel [75], may accommodate the binding of fatty-acylated substrate. Importantly, the internal cavity in the bottom of active site plays an important role in the design of isoform selective HDAC inhibitor. For example, benzamide-containing HDACi (e.g., Cpd 1 in Fig. 5D) are known to inhibit HDACs with greater isoform selectivity profiles than the hydroxamate-based inhibitors (e.g., SAHA) [74, 83]. As shown in Fig. 5, Cpd 1 interact with the fatty-acylated substrate binding site and exhibited selectivity for HDAC1/2 over other HDACs [84].
HDAC11 is the sole member of the class IV HDAC subfamily, and its biological function remains poorly understood. HDAC11 possess very weak deacetylation activity, whereas it serves as a proficient fatty acid deacylase (Fig. 5). Interestingly, HDAC11 is strongly inhibited by free fatty acids such as myristic, palmitic, and stearic acids [76]. According to the preference of HDAC11 towards longchain acyl groups, Hening Lin's group designed a new series of HDAC11 inhibitor by introducing a long hydrophobic moiety to known HDAC inhibitors [85]. The most potent HDAC11-selective inhibitor SIS17 (IC50 = 0.83 μmol/L) did not show significant inhibitory activity against other HDACs (Fig. 5E), representing the first HDAC11-selective inhibitor with cellular activity.
Most known HDACs inhibitors were substrate competitive inhibitors. Recent studies demonstrated that several HDAC isozyme possess various deacylation activities against different acylated lysine substrates. These newly discovered defatty-acylase activities of HDACs not only provide new insights into the functional effects of HDACs, but also give new opportunity for design of isoform selective inhibitor. Importantly, discover of specific HDAC inhibitor that regulating the defatty-acylation of target proteins would be valuable to understand the role of PTMs. We hope this review will be helpful to facilitate rational design of activity-guided design of HDAC selective inhibitor.
This work was supported by the National Natural Science Foundation of China (No. 81874288) and Young Scholar Research Funds of Shandong Academy of Medical Sciences (No. 2018-32).
5. Allosteric kinase inhibitors for drug discoveryJing Guo, Zhen Zhang, Xiaoyun Lu*
Small molecular kinase inhibitors (SMKIs) have become effective targeted therapeutics and shown significant clinical benefits for cancer treatment. Categorized by ATP binding pocket and DFG motif, SMKIs are classified as ATP-competitive inhibitors (reversible type I, II and irreversible type V) and allosteric inhibitors (type Ⅲ and IV) (Fig. 7). The majority of the approved SMKIs bound to the ATP-binding site are known as Type I or II [86]. However, the development of SMKIs faces two critical challenges: (I) ATP-competitive kinase inhibitors lack the relative selectivity with respect to the highly conserved ATP pocket. (II) The acquired drug resistance of SMKIs generally occurs with secondary or tertiary kinase point mutations during clinical used. Therefore, allosteric inhibitors targeting outside of the ATP pocket have recommended as a potential alternative to overcome above challenges of current SMKIs [87, 88].

|
Download:
|
| Fig. 7. The binding types of SMKIs. | |
{kind=link}
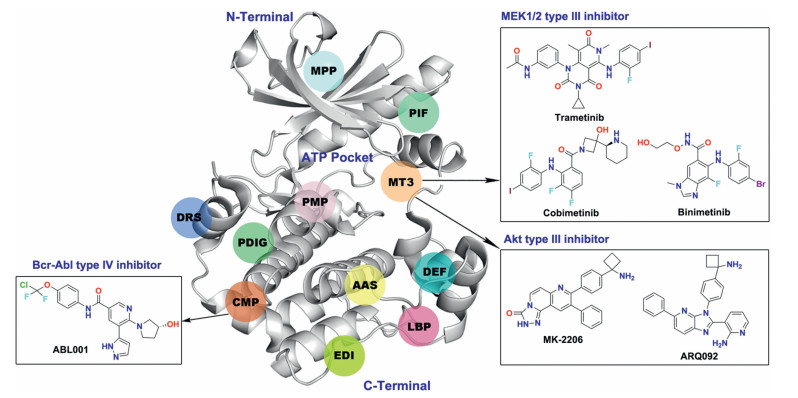
Allosteric kinase inhibitors are mainly divided into type Ⅲ and IV inhibitors based on the distance from the ATP pocket (Fig. 7). Type Ⅲ inhibitors bind to an allosteric pocket adjacent to the ATP binding pocket, such as MT3 site (Fig. 8). Type IV inhibitors bind to an allosteric site remote from the ATP binding pocket, exampled as CMP site in the N-terminal domain (Fig. 8). To date, only three allosteric inhibitors of serine/threonine kinase MEK1/2 binding to the MT3 site, cabozantinib, cobimetinib or binimetinib, have been approved as a single-agent or in combination with B-Raf inhibitors for the treatment of melanoma or non-small cancer lung cancer (Fig. 8) [89-91]. In addition, two allosteric inhibitors of serine/ threonine kinase Akt MK-2206 and ARQ092 targeting a unique allosteric site composed by MT3 site and the regulatory pleckstrin homology (PH) domain, are currently in clinical trials for the treatment of advanced solid tumors [92-95].

|
Download:
|
| Fig. 8. The reported allosteric sites of kinases and clinical drugs binding to MT3 and CMP sites. MT3: MEK1/2 Type Ⅲ inhibitor site; PIF: PDK1 interacting fragment; CMP: cAbl myristoyl pocket; DRS: D-recruitment site; DEF: docking site for ERK FXF; LBP: Lipid binding pocket; PDIG: PDIG motif site; EDI: EGFR dimerization interface; PMP: PKA myristoyl pocket; AAS: Aurora A autophosphorylation site; MPP: MKK4 p38 peptide site. | |
{kind=link}
However, none of the SMKIs approved by FDA belongs to the type IV inhibitors, but one is currently in clinical investigation, that is ABL001 (asciminib), an allosteric type IV Bcr-Abl inhibitor targeting the CMP site [96, 97]. ABL001 mimics the substrate myristate and potently binds to the CMP site of Bcr-Abl with a Kd value of 0.5–0.8 nmol/L. Furthermore, ABL001 is active in the low nanomolar concentration range against all catalytic ATP-site mutations, including the gatekeeper Bcr-Abl T315I mutation. More importantly, combination of ABL001 with the ATP-competitive inhibitor nilotinib maintains the activity against SMKIs-resistant ATP binding site mutations without shared resistance for each other. In 2019, Yueh et al. reported other sites for type IV binding mode by using Kinase Atlas, such as DRS, DEF, EDI, LBP, AAS, PDIG and PMP sites in the C-terminal domain as well as PIF and MPP sites in the N-terminal domain [98]. However, the reported allosteric inhibitors targeting these sites are still in early research, but we believe it will certainly lead to a wide range of research interests.
Overall, allosteric kinases inhibitors indeed provide greater specificity with lower adverse effects and potential to combat drug resistant mutations. In spite of the advantages offered by allosteric inhibitors, it remains some issues to be addressed. First, majority of allosteric inhibitors are combined with ATP-competitive inhibitors in vitro or in vivo, which greatly increased the difficulty of development. Second, some allosteric sites may exits such a possibility that are not essential for kinase functions compared to the ATP binding site, thus leading to incomplete kinase suppression and unfavorable anticancer activity. The other question to consider is that novel screening assays are urgently needed to identify new allosteric leads, although that numerous allosteric inhibitors have been reported in the past decade.
The authors appreciate the financial support from the National Natural Science Foundation of China (No. 81922062).
6. Structure-based design strategies of novel antiviral agents to combat drug resistanceDongwei Kang, Xinyong Liu*, Peng Zhan*
Rapid generation of drug-resistant variants is a major obstacle to current antiviral treatments. With the continued efforts in the development of increased crystallographic information on targetligand complexes and computational tools, structure-based drug discovery is a fundamental strategy for finding and optimizing antiviral agents to combat drug resistance [99-101].
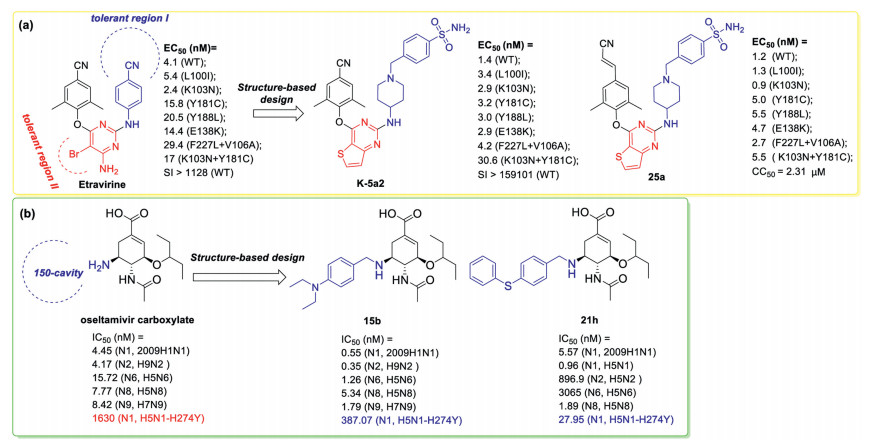
The rapid emergence of drug-resistant mutations in HIV-1 reverse transcriptase (RT) has been a key roadblock in the discovery of antiviral drugs [102, 103]. Based on the recent X-ray crystallographic studies, solvent-accessible regions including tolerant region I (the Pro236 hairpin loop) and tolerant region II (in front of Leu100, Lys101, Glu138, and Val179) in the binding site were confirmed as tolerant regions that could accommodate diverse nonnucleoside RT inhibitor (NNRTIs) [104]. Our recent optimization of etravirine targeting these two tolerant regions afforded the identification of novel anti-HIV drug candidates (exemplified by K-5a2 and 25a), which demonstrate broad spectrum of potencies against the mutant HIV-1 strains (Fig. 9a) [105-110].

|
Download:
|
| Fig. 9. Discovery of novel antiviral agents with improved potency against resistance-associated variants by structure-based optimization targeting new binding site: (a) HIV- 1 reverse transcriptase; (b) influenza virus neuraminidase. nM: nmol/L, μM: μmol/L. | |
{kind=link}
Similarly, by targeting the 150-cavity of influenza virus neuraminidase, structure-based optimization in our lab afforded novel N-substituted oseltamivir derivatives as potent anti-influenza A virus agents (exemplified by 15b and 21h) with significantly improved potency against oseltamivir-resistant N1-H274Y variant (Fig. 9b) [111, 112]. Especially, 21h was 5- to 86-fold more potent than oseltamivir carboxylate toward N1, N8, and N1-H274Y mutant neuraminidases.
Detailed analysis of the high-resolution structures of HIV-1 wild-type (WT) RT and seven RT variants bearing prevalent drugresistant mutations in complex with K-5a2 or 25a identified factors important for resilience to mutations, such as, forming the network of main chain hydrogen bonds between the NNRTIs and the binding pocket, targeting highly conserved residues in HIV-1 RT. Of particular note is that the cyanovinyl moiety of 25a is positioned in a cylindrical tunnel formed by the highly conserved Phe227 and Trp229 and makes additional interactions with this tunnel. And this may be the reason why 25a was exceptionally potent against a variety of drug-resistant HIV-1 variants compared or superior to K-5a2 (Fig. 9a) [113]. Besides, the HIV protease inhibitors are usually designed specifically to interact with protein backbone to combat drug resistance [114, 115]. These insights could be especially useful in antiviral strategies directed at newly emerging pathogens.
In current structure-based drug design, the formation of a covalent bond or hydrogen bond with a high degree of covalency [116-118], substrate envelope hypothesis [119-122], multivalent ligands [123], offer reliable strategies for combating antiviral drug resistance.
Targeted protein degradation, as a novel therapeutic modality, are playing more and more vital role in drug discovery paradigm [124]. Recently, this strategy was employed to develop a new series of Telaprevir conjugates antivirals that can both inhibit and induce proteasomal degradation of HCV NS3/4A protease. This new antiviral agents can reduce susceptibility to resistance mutations [125]. Thus, this research demonstrated that targeted protein degradation may afford a promising paradigm for the discovery of antiviral to overcome viral variants that confer resistance to traditional enzymatic inhibitors.
All in all, the acquisition of drug-resistant mutations by infectious viral pathogens remains a horrible health concern. The development of effective medicinal chemistry strategies to combat this threat is a priority. In this paper we attempt to translate the general knowledge gained from representative researches into a set of strategies to improve the antiviral drug resistance profile. Besides, multitarget-directed ligands has great potential to be effective in reducing the likelihood of drug resistance [126, 127].
Furthermore, new targets are urgently needed for the discovery of antiviral agesnt with new modes of action. For example, the multifunctional HIV-1 capsid protein [128], the indispensable ribonuclease H activity of the HIV-1 RT [129-131], are used as important, clinically unexploited therapeutic targets for drug design in our lab. Of particular concern is, host-targeted antivirals may afford an important alternative as their barriers to resistance are higher than those of agents targeting viral proteins. Also, they were also confirmed to be effective against multiple viral pathogens [132].
We acknowledge the financial support from the National Natural Science Foundation of China (NSFC, Nos. 81573347, 81973181, 81903453), Shandong Provincial Key research and development project (Nos. 2017CXGC1401, 2019JZZY021011), the Taishan Scholar Program at Shandong Province.
7. Advances in the research of nitric oxide-based anticancer agentsJianbing Wu, Yihua Zhang*, Zhangjian Huang*
Nitric oxide (NO), as a magic signaling molecule or effector in mammals, plays an important role in several biological systems, including cardiovascular, nervous, immune and other systems [133, 134]. The effects of NO in cancers depend on the type and localization of nitric oxide synthase (NOS) isoforms, concentration and duration of NO exposure, and cellular sensitivity to NO, etc. [135-138]. Generally, low levels (pmol/L-nmol/L) of NO promote cancer cell proliferation and enhance angiogenesis and metastasis, while high concentrations (μmol/L-mmol/L) of NO suppress the progression of cancer by inducing apoptosis, sensitizing tumors and reversing resistance to therapies in clinic, as well as retarding the angiogenic and metastatic cascades [135, 139-142]. RRx-001, a hypoxia-activated and epigenetic NO donor, could specifically enhance the levels of NO in cancer cells and is currently under phase II clinic trials for the treatment of solid tumor [143, 144].
With the increasing understanding of cancer biology of NO, studies in NO-based anticancer agents have achieved significant progresses, which we have comprehensively reviewed previously [138]. In this article, we briefly describe the advances made by our research group in the past three years and put forward some opinions on challenges ahead of the NO-based cancer therapy.
It is well-reported that diazeniumdiolates could be modified to be more stable with good selectivity to cancer cells via O2- protected modifications [145]. When O2-anion anchored to a group that can be removed by specific enzyme highly expressed in cancer cells, the O2-protected diazeniumdiolates could exert inhibitory effects selectively against cancer cells [146]. A great number of O2- protected diazeniumdiolates relied on specific or modified enzymatic activation has been developed and reviewed [145, 147-152]. Here, we give several examples of some compounds developed in the last years.
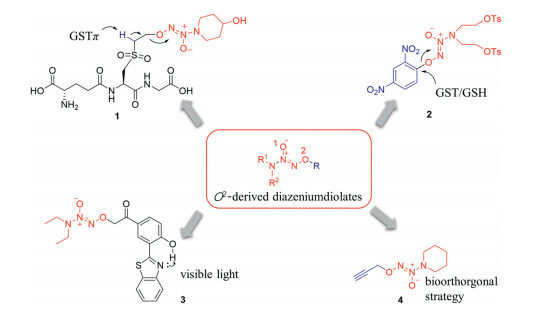
Glutathione S-transferase π (GSTπ) isoenzyme is overexpressed in many cancer cells and implicated in the development of drug resistance [153, 154]. The tyrosine (Tyr) in GSTπ is known to deprotonate the thiol group in substrate GSH, converting GSH into GS anion. A series of GSH analogues, GSH-based O2-sulfonylethyl derived diazeniumdiolates were designed and bioevaluated [155]. The representative compound 1 (Fig. 10), in which the hydrogen atom on the adjacent carbon atom of the sulfone group, could be deprotonated by the tyrosine in GSTπ in a similar way of GSTπ/GSH system. This process triggers the β-elimination reaction and subsequent liberating diazeniumdiolate anion, which can selectively release much more NO in melanoma B16 cells than that in normal epithelial cells. Moreover, 1 at 15 mg/kg significantly inhibited the growth of implanted B16 cells in nude mice by 76%.

|
Download:
|
| Fig. 10. NO-releasing small molecules triggered by the enzymes overexpressed in cancer cells and by extrinsic stimulus. | |
{kind=link}
Some diazeniumdiolate-based NO donors triggered by other specific enzymes or extrinsic stimulus were synthesized and biologically evaluated recently. For instance, O2-(2, 4-dinitrophenyl) derived diazeniumdiolate 2 [156] are GSTπ/GSH activatable, which selectively generated NO and suppressed proliferation of human leukemia cells. O2-(3-(benzothiazole-2-yl)-4-hydroxyphenacyl)diazeniumdiolate 3 is ultraviolet light responsive and acted as a photosensitizer capable to release NO in a real-time controllable and monitorable manner in living cancer cells [157]. Furthermore, O2-alkyl derived diazeniumdiolate 4 is "bondcleavable" responsive, in which a biorthogonal chemistry is involved [158]. This extrinsic stimulus strategy would expand the therapeutic potential of NO-based anticancer agents in the near future.
Apart from improving selectivity to cancer cells versus normal ones, the more important issue in the research of NO donor-based anticancer agents is how to targetedly deliver the agents into cancer cells or tissues. Enhanced permeability and retention (EPR) effect of nanoparticles could be taken advantage to solve this problem. Giles et al. summarized NO-donating nanoparticles or macromolecules where polymeric scaffolds, including microbeads, dendrimers, gold nanoparticles, zeolites, metal-organic framework, etc. have been utilized in recent years [159-161].
With the cooperation with materials chemists, a redox/ enzyme-responsive NO-donating nanoparticle was designed and synthesized recently [162]. Briefly, a disulfide-doped and organic– inorganic hybrid nanocarrier (PEGylated disulfide-doped hybrid nanocarriers, PDHNs) encapsulated GSTπ/GSH responsive NO donating compound NPQ and an aggregation-induced-emission red fluorogen QM-2 to construct a redox/enzyme-responsive nanoparticle QM-NPQ@PDHNs (Fig. 11A). The nanoparticle could be stable in in vivo circulation and accumulated in tumor tissue by EPR effect. Owing to the high concentrations of GSH in cancer cells, biodegradation of disulfide-containing PDHN was triggered before subsequent NO release in situ. It was observed that the nanoparticle had little influence on the blood pressure as compared to small molecule NPQ. And this nanoparticle indeed accumulated in tumor tissues verse normal ones, which can be monitored by three-dimensional florescence images. Importantly, the nanoparticle inhibited the cancer cell growth by 84% (w/w), higher than small molecule NPQ (49%) at the same dose.

|
Download:
|
| Fig. 11. Release of NO from (A) nanoparticles, (B) nano-coordinatin polymer and (C) antibody-NO conjugates. | |
{kind=link}
In order to avoid the accumulation toxicity of nanocarrier, and to enhance the druggability of NO-donating nanoparticle, a novel carrier-free ferrum(II)-NO nanoscale coordination polymer (FNCP) was designed and synthesized (Fig. 11B), which is comprised of GSTπ/GSH responsive NO donors with terminal carboxyl groups coordinating with ferrous ions [163]. The polymer with dimeter of approximately 88 nm was stable in the circulation and could be accumulated in the tumor site in vivo, where it generated high levels of NO and reactive oxygen species (ROS). Indeed, ferrous ions in FNCP as a chemodynamic therapy (CDT) component acted synergistically with NO to inhibit the growth of liver cancer with high efficacy and safety.
Recently, monoclonal antibody drugs with high target recognition, affinity and internalization rates have been attracted much attention in the field of cancer therapy. However, the antibody drugs faces with the issues of insufficient therapeutic efficacy and narrow therapeutic window. Antibody drug conjugates (ADCs) has been therefore designed, acting as smart missiles carrying around with cytotoxins targeting cancer cells and thus displaying better anticancer effects and enhanced therapeutic windows [164, 165].
Encouraged by the concept of ADCs, we conjugated specific monoclonal antibody with NO donors to produce a new class of antibody-NO conjugates (ANCs) [166]. HN-01 was prepared by conjugation of a NO donor bearing a GSH-responsive disulfide bond with an antibody G7mAb (Fig. 11C). HN-01 was selectively enriched on the surface of hepatic carcinoma CD24 cells, thus improving the therapeutic index of NO with little side effects on normal tissues. This study is the first to expand the concept of ADCs for spatiotemporal controlled release of NO, and this class of ANCs could have desirable druggability warranting further investigations.
Due to the diverse biological functions of NO, the precise control of amount and localization of NO release is crucial for NO-based cancer therapy. In general, amount-control may be achieved by employing different NO donors with different behaviors. For instance, diazeniumdiolate compounds can spontaneously or quickly release high levels of NO while nitrate esters are generally able to slowly release small amounts of NO. As for localizationcontrol, environmental stimulation, targeting delivery, topical administration or utilizing precursor of NO such as arginine or organic nitrite could be exploited.
Importantly, new mechanism of anticancer action of NO should be intensively investigated. Over the years, researchers have been particularly interested in exploration of protein nitration and Snitrosation mediated by NO donors. Protein tyrosine nitration is an irreversible modification by nitrating agents such as peroxynitrite, which derived from the reaction of endogenic or exogenic NO with superoxide anion (O2·-) [167]. Nitration in the framework of PAK1 signaling and heat shock protein 90 (Hsp90) in cancer cells have been documented [168, 169]. The interaction between the nitrating product 3-nitrotyrosine and 8-nitroguanine DNA, inducing apoptosis of cancer cell, has been reported [170]. Similarly, protein S-nitrosation, as a common post-translational modification, is an important regulatory mechanism in carcinogenesis and metastasis, which may result in the changes of the target protein activity and function, displaying the same important roles in physiology and pathology as phosphorylation and acetylation [171]. Based on these mechanisms, protein S-nitrosation and nitrosation probes and proteomic approaches are urgently needed to reveal the underlying molecular mechanisms for those NO-donating anticancer agents. Notably, real-time NO measurement in cancer tissues in living animals after administration of NO donor agents remains a great challenge, making an unclear concentrationactivity relationship of NO in cancer biology.
We have reason to believe that, with the development of life science and information science disciplines, cooperation of medicinal chemists with molecular biologists in NO donor-based cancer therapy will make more and more progresses.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81822041, 81773573, 21977116, and 81673305), National Science & Technology Major Project "Key New Drug Creation and Manufacturing Program" (No.2018ZX09711002-006-013), the open projects from State Key Laboratory of Pathogenesis, Prevention and Treatment of High Incidence Diseases in Central Asia (No. SKL-HIDCA-2018-1), and State Key Laboratory of Natural Medicines (No.SKLNMZZCX201824), part of the work was supported by Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX18_0795).
8. Photoactivation strategies in medicinal chemistryZhihong Li, Xiaojin Zhang*
The majority of current medical treatments rely on the use of small bioactive molecules, which invoke bioactivity by interacting with biomolecular targets. Thus, the selectivity of this interaction is crucial, and the lack thereof can lead to various clinical side effects. Recently, the use of photopharmacology has aimed at solving the problems of off-target effects, by establishing biocompatible photoactivatable systems for controlling the action of small molecules. This is achieved by the use of light to activate the biological functions of target compounds. Current studies have found that light offers more precise spatiotemporal control of the activation step, as compared with other external stimuli. Hence, the use of photoactivatable molecules could overcome environmental and systemic side effects of drugs, via the selective activation of bioactivity upon irradiation with appropriate wavelengths of light in order to induce desired therapeutic effects [172, 173].
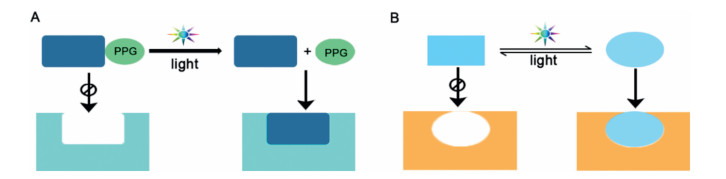
Photopharmacology has grown into a vibrant field in the last decade, leading to a wide range of applications in current medicinal research [174]. As depicted in Fig. 12, two molecular strategies have been extensively developed for controlling chemical and biological processes with light [175]. The first strategy relies on caging the functional group of an active molecule with a photoremovable protecting group (PPG) and then liberating its functionality using light. The second strategy is concerned with molecular photoswitches that can be reversibly isomerized between two or more states upon photoactivation. Such isomerization results in an alteration in molecular properties, which in some cases will be translated into a change in the biological effect [176]. This review will summarize and provide insight into recent research advances in the understanding of photoactivatable caged prodrugs and photoswitchable molecules.

|
Download:
|
| Fig. 12. Basic principles and requirements for light-controlled treatment modalities. (A) photoactivatable caged prodrugs, (B) reversible photoactivation/inactivation using a photochromic ligand that toggles between inactive and active form. | |
{kind=link}
Photoactivatable caged prodrugs represent a new strategy in modulating the action of bioactive compounds in a spatially and temporally controlled manner, employing a PPG. The PPG is a chromophore that covalently links to the pharmacophoric group of the bioactive compound, thus inhibiting its bioactivity. This method of action is known as "caging" of the photoactivatable prodrug. The covalent bond between the PPG and the bioactive compound is cleaved upon irradiation with light, resulting in the liberation of the bioactive molecule. Over the past decade, there has been an upsurge of interest in PPGs that has resulted in a dramatic increase in the number of photosensitive and biocompatible scaffolds, making them suitable for biological applications. Simplified representative structures (Z1-Z8) of the currently available PPGs are listed in Fig. 13 [175].

|
Download:
|
| Fig. 13. Representation of the different PPGs, where R is the caged group (red). | |
{kind=link}
In recent years, the off-target effects of systemically administered anticancer drugs have heavily constrained their efficacy and tolerability. The photoactivatable prodrug concept has been applied to reduce these off-target effects. To ultimately conquer the severe side effects and drug resistance of the anti-melanoma agent vemurafenib, Horbert et al. have incorporated PPGs into the drug (Fig. 14) [177]. By targeted irradiation of vermurafenib caged with kinase inhibitor (PPG-9), vermurafenib (9) can be released at high concentration in a precisely controlled way, and only at the sites afflicted by disease. Moreover, PPG-idasanutlin (PPG-10), a photoactivatable murine double minute 2 (MDM2) inhibitor, exerts no functional effect on cellular outgrowth, but allows for the selective, noninvasive activation of anti-tumor properties upon irradiation at 400 nm [178]. Recently, Li et al. have reported the first application of the photoactivatable prolyl hydroxylase domain 2 (PHD2) inhibitor (PPG-11). The inhibitory activity of PHD2 can be controlled by 405 nm irradiation, subsequently stabilizing hypoxia inducible factor (HIF) and promoting expression of the target gene [179]. Currently, as with photoactivatable prodrugs, many new discoveries have brought substantial improvements and versatility to the absorption properties, as well as improved efficiencies and rates of release [180]. Nonetheless, many vital research goals still remain, such as extending the wavelength coverage for the activation of PPGs into the infrared region, and improving the selective absorption at certain wavelengths. The authors consider that photoactivatable caged prodrugs, although not yet at the clinical development stage, have the potential to become an important clinical application of light to medicine, since it could lead to the photocontrolled and selective addressing of targets in the human body by photoactivatable small molecules [181].

|
Download:
|
| Fig. 14. Representative photoactivatable caged inhibitors. | |
{kind=link}
Photoswitchable molecules, also termed photochromic compounds, are a class of molecules whose activity at a target of interest can be controlled precisely, and in a reversible manner, by light. Photoswitchable compounds, such as azobenzene (12 and 13), stilbene (14 and 15), diarylethene (16 and 17), and spiropyran (18 and 19), exhibit reversible photoisomerization upon irradiation with light (Fig. 15A). When irradiated with light of a particular wavelength, such molecules undergo a reversible configurational change in their structure, which may substantially alter their binding affinity to specific targets. Among these molecules, azobenzene is to date the most extensively used photoswitch for biological applications, due to its favorable geometric and photochemical properties [182]. It is mainly applied to ion channels and G protein-coupled receptors (GPCRs). Grutter and colleagues reported that purinergic receptor (P2X) channels, covalently modified by azobenzene-containing reagents (20 and 21, Fig. 15B) at the transmembrane segments, could be reversibly turned on and off upon irradiation with light, without need for the natural ligand ATP [183]. These light-gated P2X receptors represent valuable tools for investigating the functions of P2X receptor. Hauwert et al. developed a bidirectional antagonist toolbox for the histamine H3 receptor (H3R). UF14738 (22 and 23) and VUF14862 (24 and 25) were demonstrated to swiftly and reversibly photoisomerize, yielding a more than 10-fold increase and decrease in H3R binding affinity, respectively, upon illumination at 360 nm. Both ligands are robust and fatigueresistant photoswitchable GPCR antagonists, making them suitable for spatiotemporal studies of H3R signaling [184]. Moreover, VUF16216 (26 and 27) is the GPCR azo-ligand switch of the highest photoinducing efficacy reported to date, and provides effective, real-time switching from antagonism to agonism [185]. The special properties of azobenzene-based photoswitches enable powerful and reversible light-control of biological functions. Currently, an important goal in the development of azobenzene-based photoswitches is their application in vivo. It is notable that UV light is needed to trigger azobenzene photoswitches from trans to cis isomer, however, UV light is ineffective in penetrating the skin and tissue. Therefore, photoswitches utilizing longer wavelengths for trans to cis isomerization are desired.

|
Download:
|
| Fig. 15. (A) Overview of the most commonly used photoswitches discussed in this review and their switching characteristics. (B) Representatives of the azobenzene class of photoswitchable molecules. | |
{kind=link}
In general, it is a painstaking journey from the design of photoactivatable molecules to their clinical application, but the ultimate reward will be innovative approaches to precisely regulate biological processes with light.
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81973173 and 81773571), Jiangsu Province Funds for Excellent Young Scientists (No.BK20170088), the Six Talent Peaks Project (No. YY-023) and the 333 Project of Jiangsu Province.Declaration of competing interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
| [1] |
D. Plewczynski, M. Lazniewski, R. Augustyniak, K. Ginalski, J. Comput. Chem. 32 (2011) 742-755. DOI:10.1002/jcc.21643 |
| [2] |
I.D. Kuntz, J.M. Blaney, S.J. Oatley, R. Langridge, T.E. Ferrin, J. Mol. Bio. 161 (1982) 269-288. DOI:10.1016/0022-2836(82)90153-X |
| [3] |
Z. Zsoldos, D. Reid, A. Simon, S.B. Sadjad, A.P. Johnson, J. Mol. Graph. Model 26 (2007) 198-212. DOI:10.1016/j.jmgm.2006.06.002 |
| [4] |
M.K. Rarey, B. Kramer, T. Lengauer, G. Klebe, J. Mol. Bio. 261 (1996) 470-489. DOI:10.1006/jmbi.1996.0477 |
| [5] |
R.A. Friesner, J.L. Banks, R.B. Murphy, et al., J. Med. Chem. 47 (2004) 1739-1749. DOI:10.1021/jm0306430 |
| [6] |
G.W. Jones, P. Willete, R.C. Glen, A.R. Leach, R. Taylor, J. Mol. Bio. 267 (1997) 727-748. DOI:10.1006/jmbi.1996.0897 |
| [7] |
C. Venkatachalam, X. Jiang, T. Oldfield, M. Waldman, J. Mol. Graphics Modell. 21 (2003) 289-307. DOI:10.1016/S1093-3263(02)00164-X |
| [8] |
A.N. Jain, J. Med. Chem. 46 (2003) 499-511. DOI:10.1021/jm020406h |
| [9] |
G.M. Morris, R. Huey, W. Lindstrom, et al., J. Comput. Chem. 30 (2009) 2785-2791. DOI:10.1002/jcc.21256 |
| [10] |
L. Rao, B. Chi, Y. Ren, et al., J. Comput. Chem. 37 (2016) 336-344. DOI:10.1002/jcc.24217 |
| [11] |
L. Wei, B. Chi, Y. Ren, et al., J. Chem. Theory Comput. 15 (2019) 4264-4279. DOI:10.1021/acs.jctc.8b01150 |
| [12] |
W. Guo, A. Wu, I.Y. Zhang, X. Xu, J. Comput. Chem. 33 (2012) 2142-2160. DOI:10.1002/jcc.23051 |
| [13] |
M. Zheng, X. Liu, Y. Xu, et al., Trends Pharmacol. Sci. 34 (2013) 549-559. DOI:10.1016/j.tips.2013.08.004 |
| [14] |
S.Y. Yang, Drug Discov. Today 15 (2010) 444-450. DOI:10.1016/j.drudis.2010.03.013 |
| [15] |
X. Liu, H. Jiang, H. Li, J. Chem. Inf. Model. 51 (2011) 2372-2385. DOI:10.1021/ci200060s |
| [16] |
W. Lu, X. Liu, X. Cao, et al., J. Med. Chem. 54 (2011) 3564-3574. DOI:10.1021/jm200139j |
| [17] |
G.B. Li, S. Ji, L.L. Yang, et al., Eur. J. Med. Chem. 93 (2015) 523-538. DOI:10.1016/j.ejmech.2015.02.019 |
| [18] |
G.B. Li, M.I. Abboud, J. Brem, et al., Chem. Sci. 8 (2017) 928-937. DOI:10.1039/C6SC04524C |
| [19] |
K.A. O'Connor, B.L. Roth, Nat. Rev. Drug Discov. 4 (2005) 1005-1014. DOI:10.1038/nrd1900 |
| [20] |
A. Corbett, J. Pickett, A. Burns, et al., Nat. Rev. Drug Discov. 11 (2012) 833-846. DOI:10.1038/nrd3869 |
| [21] |
Y.Z. Chen, D.G. Zhi, Proteins 43 (2001) 217-226. DOI:10.1002/1097-0134(20010501)43:2<217::AID-PROT1032>3.0.CO;2-G |
| [22] |
H. Li, Z. Gao, L. Kang, et al., Nucleic Acids Res. 34 (2006) W219-W224. DOI:10.1093/nar/gkl114 |
| [23] |
J.C. Wang, P.Y. Chu, C.M. Chen, J.H. Lin, Nucleic Acids Res. 40 (2012) W393-W399. DOI:10.1093/nar/gks496 |
| [24] |
K.T. Schomburg, S. Bietz, H. Briem, et al., J. Chem. Inf. Model. 54 (2014) 1676-1686. DOI:10.1021/ci500130e |
| [25] |
X. Liu, S. Ouyang, B. Yu, et al., Nucleic Acids Res. 38 (2010) W609-W614. DOI:10.1093/nar/gkq300 |
| [26] |
X. Wang, Y. Shen, S. Wang, et al., Nucleic Acids Res. 45 (2017) W356-W360. DOI:10.1093/nar/gkx374 |
| [27] |
A. Brakoulias, R.M. Jackson, Proteins 56 (2004) 250-260. DOI:10.1002/prot.20123 |
| [28] |
C. Schalon, J.S. Surgand, E. Kellenberger, D. Rognan, Proteins 71 (2008) 1755-1778. DOI:10.1002/prot.21858 |
| [29] |
Z. Chen, X. Zhang, C. Peng, et al., J. Chem. Inf. Model. 59 (2019) 3353-3358. DOI:10.1021/acs.jcim.9b00332 |
| [30] |
M.J. Keiser, B.L. Roth, B.N. Armbruster, et al., Nat. Biotechnol. 25 (2007) 197-206. DOI:10.1038/nbt1284 |
| [31] |
M.J. Keiser, V. Setola, J.J. Irwin, et al., Nature 462 (2009) 175-181. DOI:10.1038/nature08506 |
| [32] |
J. Gong, C. Cai, X. Liu, et al., Bioinformatics 29 (2013) 1827-1829. DOI:10.1093/bioinformatics/btt270 |
| [33] |
S.L. Kinnings, R.M. Jackson, J. Chem. Inf. Model. 51 (2011) 624-634. DOI:10.1021/ci1003174 |
| [34] |
G.B. Li, L.L. Yang, Y. Xu, et al., J. Mol. Graph. Model. 44 (2013) 278-285. DOI:10.1016/j.jmgm.2013.07.005 |
| [35] |
G.B. Li, Z.J. Yu, S. Liu, et al., J. Chem. Inf. Model. 57 (2017) 1640-1651. DOI:10.1021/acs.jcim.7b00225 |
| [36] |
G.B. Li, L.L. Yang, W.J. Wang, et al., J. Chem. Inf. Model. 53 (2013) 592-600. DOI:10.1021/ci300493w |
| [37] |
X. Zhou, M. Wu, Y. Xie, et al., Front. Pharmacol. 9 (2018) 1238. DOI:10.3389/fphar.2018.01238 |
| [38] |
Z. Li, X. Li, X. Liu, et al., Bioinformatics 35 (2019) 5354-5356. DOI:10.1093/bioinformatics/btz519 |
| [39] |
D.J. Klionsky, S.D. Emr, Science 290 (2000) 1717-1721. DOI:10.1126/science.290.5497.1717 |
| [40] |
R. Khandia, M. Dadar, A. Munjal, et al., Cells 8 (2019) E674. DOI:10.3390/cells8070674 |
| [41] |
N. Wang, L. Wang, X.Q. Xie, J. Chem. Inf. Model. 57 (2017) 2686-2698. DOI:10.1021/acs.jcim.7b00277 |
| [42] |
T. Xie, L. Zhang, S. Zhang, et al., Oncotarget 7 (2016) 10015-10022. DOI:10.18632/oncotarget.7015 |
| [43] |
A.C. Jacomin, S. Samavedam, V. Promponas, I.P. Nezis, Autophagy 12 (2016) 1945-1953. DOI:10.1080/15548627.2016.1207016 |
| [44] |
Y. Deng, L. Zhu, H. Cai, et al., Cell. Proliferat. 51 (2018) e12403. DOI:10.1111/cpr.12403 |
| [45] |
N.N. Wang, J. Dong, L. Zhang, et al., J. Cheminform. 10 (2018) 34. DOI:10.1186/s13321-018-0289-4 |
| [46] |
R. Nanduri, R. Kalra, E. Bhagyaraj, et al., Autophagy 15 (2019) 1280-1295. DOI:10.1080/15548627.2019.1571717 |
| [47] |
C.S. Börlin, V. Lang, A. Hamacher-Brady, N.R. Brady, Cell Commun. Signal 12 (2014) 56.
|
| [48] |
I. Tavassoly, J. Parmar, A.N. Shajahan-Haq, et al., CPT Pharmacometrics Syst. Pharmacol. 4 (2015) 263-272. DOI:10.1002/psp4.29 |
| [49] |
J. Scheidel, L. Amstein, J. Ackermann, et al., PLoS Comput. Biol. 12 (2016) e1005200. DOI:10.1371/journal.pcbi.1005200 |
| [50] |
E. Aslan, N. Küçükoglu, M. Arslanyolu, Peer J 5 (2017) e2878. DOI:10.7717/peerj.2878 |
| [51] |
F.M. Awan, A. Obaid, A. Ikram, H.A. Janjua, Int. J. Mol. Sci. 18 (2017) E139. DOI:10.3390/ijms18010139 |
| [52] |
T.E. Hoffman, K.J. Barnett, L. Wallis, W.H. Hanneman, Aging Cell 16 (2017) 1244-1255. DOI:10.1111/acel.12644 |
| [53] |
J. Zhou, D. Hang, Y. Jiang, et al., Gene 627 (2017) 549-555. DOI:10.1016/j.gene.2017.06.053 |
| [54] |
J.M. Heo, A. Ordureau, S. Swarup, et al., Sci. Adv. 4 (2018) eaav0443. DOI:10.1126/sciadv.aav0443 |
| [55] |
C. Xuan, Y. Gao, M. Jin, et al., IUBMB Life 71 (2019) 827-834. DOI:10.1002/iub.1999 |
| [56] |
J. Pei, N. Yin, X. Ma, L. Lai, J. Am. Chem. Soc. 136 (2014) 11556-11565. DOI:10.1021/ja504810z |
| [57] |
S. Lu, M. Ji, D. Ni, J. Zhang, Drug Discov. Today 23 (2018) 359-365. DOI:10.1016/j.drudis.2017.10.001 |
| [58] |
X. Yang, Y. Wang, R. Byrne, et al., Chem. Rev. 119 (2019) 10520-10594. DOI:10.1021/acs.chemrev.8b00728 |
| [59] |
X.J. Yang, E. Seto, Oncogene 26 (2007) 5310-5318. DOI:10.1038/sj.onc.1210599 |
| [60] |
G.P. Delcuve, D.H. Khan, J.R. Davie, Clin. Epigenetics 4 (2012) 5. DOI:10.1186/1868-7083-4-5 |
| [61] |
P. Bheda, H. Jing, C. Wolberger, et al., Annu. Rev. Biochem. 85 (2016) 405-429. DOI:10.1146/annurev-biochem-060815-014537 |
| [62] |
M.D. Resh, Prog. Lipid Res. 63 (2016) 120-131. DOI:10.1016/j.plipres.2016.05.002 |
| [63] |
H. Jiang, X.Y. Zhang, H.N. Lin, Sci. Rep. 6 (2016) 24371.
|
| [64] |
T. Peng, E. Thinon, H.C. Hang, Curr. Opin. Chem. Biol. 30 (2016) 77-86. DOI:10.1016/j.cbpa.2015.11.008 |
| [65] |
J.L. Feldman, J. Baeza, J.M. Denu, J. Biol. Chem. 288 (2013) 31350-31356. DOI:10.1074/jbc.C113.511261 |
| [66] |
J.L. Feldman, K.E. Dittenhafer-Reed, N. Kudo, et al., Biochemistry 54 (2015) 3037-3050. DOI:10.1021/acs.biochem.5b00150 |
| [67] |
A.S. Madsen, C. Andersen, M. Daoud, et al., J. Biol. Chem. 291 (2016) 7128-7141. DOI:10.1074/jbc.M115.668699 |
| [68] |
X.Y. Zhang, S. Khan, H. Jiang, et al., Nat. Chem. Biol. 12 (2016) 614-620. DOI:10.1038/nchembio.2106 |
| [69] |
H. Jiang, S. Khan, Y. Wang, et al., Nature 496 (2013) 110-113. DOI:10.1038/nature12038 |
| [70] |
Z. Tong, Y. Wang, X.Y. Zhang, et al., ACS Chem. Biol. 11 (2016) 742-747. DOI:10.1021/acschembio.5b01084 |
| [71] |
Z. Tong, M. Wang, Y. Wang, et al., ACS Chem. Biol. 12 (2017) 300-310. DOI:10.1021/acschembio.6b00954 |
| [72] |
P. Aramsangtienchai, N.A. Spiegelman, B. He, et al., ACS Chem. Biol. 11 (2016) 2685-2692. DOI:10.1021/acschembio.6b00396 |
| [73] |
C. Moreno-Yruela, I. Galleano, A.S. Madsen, et al., Cell Chem. Biol. 25 (2018) 849-856. DOI:10.1016/j.chembiol.2018.04.007 |
| [74] |
M.K. Wambua, D.A. Nalawansha, A.T. Negmeldin, et al., J. Med. Chem. 57 (2014) 642-650. DOI:10.1021/jm401837e |
| [75] |
D.F. Wang, O. Wiest, P. Helquist, et al., J. Med. Chem. 47 (2004) 3409-3417. DOI:10.1021/jm0498497 |
| [76] |
Z. Kutil, Z. Novakova, M. Meleshin, et al., ACS Chem. Biol. 13 (2018) 685-693. DOI:10.1021/acschembio.7b00942 |
| [77] |
L.L. Yang, X.B. Ma, C. Yuan, et al., Eur. J. Med. Chem. 134 (2017) 230-241. DOI:10.1016/j.ejmech.2017.04.010 |
| [78] |
L.L. Yang, H.L. Wang, L. Zhong, et al., Eur. J. Med. Chem. 155 (2018) 806-823. DOI:10.1016/j.ejmech.2018.06.041 |
| [79] |
B.C. Smith, J.M. Denu, Biochemistry 46 (2007) 14478-14486. DOI:10.1021/bi7013294 |
| [80] |
H. Jing, J. Hu, B. He, et al., Cancer Cell 29 (2016) 767-768. DOI:10.1016/j.ccell.2016.04.005 |
| [81] |
N.A. Spiegelman, J.Y. Hong, J. Hu, et al., ChemMedChem 14 (2019) 744-748. DOI:10.1002/cmdc.201800715 |
| [82] |
A.S. Farooqi, J.Y. Hong, J. Cao, et al., J. Med. Chem. 62 (2019) 4131-4141. DOI:10.1021/acs.jmedchem.9b00191 |
| [83] |
L. Whitehead, M.R. Dobler, B. Radetich, et al., Bioorg. Med. Chem. 19 (2011) 4626-4634. DOI:10.1016/j.bmc.2011.06.030 |
| [84] |
B.E.L. Lauffer, R. Mintzer, R.N. Fong, et al., J. Biol. Chem. 288 (2013) 26926-26943. DOI:10.1074/jbc.M113.490706 |
| [85] |
S.I. Son, J. Cao, C.L. Zhu, et al., ACS Chem. Biol. 14 (2019) 1393-1397. DOI:10.1021/acschembio.9b00292 |
| [86] |
R. Roskoski Jr., Pharmacol. Res. 144 (2019) 19-50. DOI:10.1016/j.phrs.2019.03.006 |
| [87] |
P. Wu, M.H. Clausen, T.E. Nielsen, Pharmacol. Therap. 156 (2015) 59-68. DOI:10.1016/j.pharmthera.2015.10.002 |
| [88] |
S.W. Cowan-Jacob, W. Jahnke, S. Knapp, Future Med. Chem. 6 (2014) 541-561. DOI:10.4155/fmc.13.216 |
| [89] |
H. Abe, S. Kikuchi, K. Hayakawa, et al., ACS Med. Chem. Lett. 2 (2011) 320-324. DOI:10.1021/ml200004g |
| [90] |
K.D. Rice, N. Aay, N.K. Anand, et al., ACS Med. Chem. Lett. 3 (2012) 416-421. DOI:10.1021/ml300049d |
| [91] |
A.A.N. Rose, Drugs Today (Barc) 55 (2019) 247-264. DOI:10.1358/dot.2019.55.4.2958476 |
| [92] |
C. Robert, B. Karaszewska, J. Schachter, et al., N. Engl. J. Med. 372 (2015) 30-39. DOI:10.1056/NEJMoa1412690 |
| [93] |
H. Hirai, H. Sootome, Y. Nakatsuru, et al., Mol. Cancer Therap. 9 (2010) 1956-1967. DOI:10.1158/1535-7163.MCT-09-1012 |
| [94] |
T.A. Yap, L. Yan, A. Patnaik, et al., Clin. Cancer Res. 20 (2014) 5672-5685. DOI:10.1158/1078-0432.CCR-14-0868 |
| [95] |
J.M. Lapierre, S. Eathiraj, D. Vensel, et al., J. Med. Chem. 59 (2016) 6455-6469. DOI:10.1021/acs.jmedchem.6b00619 |
| [96] |
A.A. Wylie, J. Schoepfer, W. Jahnke, et al., Nature 543 (2017) 733-737. DOI:10.1038/nature21702 |
| [97] |
J. Schoepfer, W. Jahnke, G. Berellini, et al., J. Med. Chem. 61 (2018) 8120-8135. DOI:10.1021/acs.jmedchem.8b01040 |
| [98] |
C. Yueh, J. Rettenmaier, B. Xia, et al., J. Med. Chem. 62 (2019) 6512-6524. DOI:10.1021/acs.jmedchem.9b00089 |
| [99] |
E. de Clercq, Med. Res. Rev. 33 (2013) 1249-1277. DOI:10.1002/med.21281 |
| [100] |
P. Zhan, C. Pannecouque, E. de Clercq, X. Liu, J. Med. Chem. 59 (2016) 2849-2878. DOI:10.1021/acs.jmedchem.5b00497 |
| [101] |
X. Zuo, Z. Huo, D. Kang, et al., Expert Opin. Ther. Pat. 28 (2018) 299-316. DOI:10.1080/13543776.2018.1438410 |
| [102] |
P. Zhan, X. Chen, D. Li, et al., Med. Res. Rev. 33 (2013) E1-E72. |
| [103] |
X. Jiang, J. Yu, Z. Zhou, et al., Med. Res. Rev. 39 (2019) 2194-2238. DOI:10.1002/med.21581 |
| [104] |
B. Huang, W. Chen, T. Zhao, et al., J. Med. Chem. 62 (2019) 2083-2098. DOI:10.1021/acs.jmedchem.8b01729 |
| [105] |
D. Kang, H. Zhang, Z. Wang, et al., J. Med. Chem. 62 (2019) 1484-1501. DOI:10.1021/acs.jmedchem.8b01656 |
| [106] |
D. Kang, Z. Fang, B. Huang, et al., J. Med. Chem. 60 (2017) 4424-4443. DOI:10.1021/acs.jmedchem.7b00332 |
| [107] |
D. Kang, Z. Fang, Z. Li, et al., J. Med. Chem. 59 (2016) 7991-8007. DOI:10.1021/acs.jmedchem.6b00738 |
| [108] |
D.W. Kang, T. Zhao, Z. Wang, et al., Commun. Chem. 2 (2019) 74.
|
| [109] |
Z. Huo, H. Zhang, D. Kang, et al., ACS Med. Chem. Lett. 9 (2018) 334-338. DOI:10.1021/acsmedchemlett.7b00524 |
| [110] |
D. Kang, F.X. Ruiz, D. Feng, et al., J. Med. Chem. 63 (2020) 1298-1312. DOI:10.1021/acs.jmedchem.9b01769 |
| [111] |
J. Zhang, V. Poongavanam, D. Kang, et al., J. Med. Chem. 61 (2018) 6379-6397. DOI:10.1021/acs.jmedchem.8b00929 |
| [112] |
J. Zhang, N.A. Murugan, Y. Tian, et al., J. Med. Chem. 61 (2018) 9976-9999. DOI:10.1021/acs.jmedchem.8b01065 |
| [113] |
Y. Yang, D. Kang, L.A. Nguyen, et al., eLife 7 (2018) e36340. DOI:10.7554/eLife.36340 |
| [114] |
A.K. Ghosh, B.D. Chapsal, I.T. Weber, H. Mitsuya, Acc.Chem. Res. 41 (2008) 78-86. DOI:10.1021/ar7001232 |
| [115] |
A.K. Ghosh, D.D. Anderson, I.T. Weber, H. Mitsuya, Angew. Chem. Int. Ed. 51 (2012) 1778-1802. DOI:10.1002/anie.201102762 |
| [116] |
G. Wu, T. Zhao, D. Kang, et al., J. Med. Chem. 62 (2019) 9375-9414. DOI:10.1021/acs.jmedchem.9b00359 |
| [117] |
I.W. Windsor, M.J. Palte, J.C. Lukesh 3rd., et al., J. Am. Chem. Soc. 140 (2018) 14015-14018. DOI:10.1021/jacs.8b07366 |
| [118] |
A.H. Chan, W.G. Lee, K.A. Spasov, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 9725-9730. DOI:10.1073/pnas.1711463114 |
| [119] |
M.D. Altman, A. Ali, G.S. Reddy, et al., J. Am. Chem. Soc. 130 (2008) 6099-6113. DOI:10.1021/ja076558p |
| [120] |
L.N. Rusere, G.J. Lockbaum, S.K. Lee, et al., J. Med. Chem. 62 (2019) 8062-8079. DOI:10.1021/acs.jmedchem.9b00838 |
| [121] |
X.Z. Zhao, S.J. Smith, D.P. Maskell, et al., J. Med. Chem. 60 (2017) 7315-7332. DOI:10.1021/acs.jmedchem.7b00596 |
| [122] |
A.N. Matthew, J. Zephyr, C.J. Hill, et al., J. Med. Chem. 60 (2017) 5699-5716. DOI:10.1021/acs.jmedchem.7b00426 |
| [123] |
Y. Song, P. Zhan, X. Li, et al., Curr. Med. Chem. 20 (2013) 815-832. |
| [124] |
D.P. Bondeson, A. Mares, I.E. Smith, et al., Nat. Chem. Biol. 11 (2015) 611-617. DOI:10.1038/nchembio.1858 |
| [125] |
M. de Wispelaere, G. Du, K.A. Donovan, et al., Nat. Commun. 10 (2019) 3468.
|
| [126] |
P. Zhan, X. Liu, Curr. Pharm. Des. 15 (2009) 1893-1917. DOI:10.2174/138161209788453266 |
| [127] |
P. Zhan, X. Liu, Curr. Med. Chem. 20 (2013) 1743-1758. DOI:10.2174/0929867311320130011 |
| [128] |
G. Wu, W.A. Zalloum, M.E. Meuser, et al., Eur. J. Med. Chem. 158 (2018) 478-492. DOI:10.1016/j.ejmech.2018.09.029 |
| [129] |
L. Sun, P. Gao, G. Dong, et al., Eur. J. Med. Chem. 155 (2018) 714-724. DOI:10.1016/j.ejmech.2018.06.036 |
| [130] |
P. Gao, X. Wang, L. Sun, et al., Chem. Biol. Drug Des. 93 (2019) 582-589. DOI:10.1111/cbdd.13455 |
| [131] |
L. Wang, S.G. Sarafianos, Z. Wang, Acc. Chem. Res. 53 (2020) 218-230. DOI:10.1021/acs.accounts.9b00450 |
| [132] |
Y. Yang, L. Cao, H. Gao, et al., J. Med. Chem. 62 (2019) 4056-4073. DOI:10.1021/acs.jmedchem.9b00091 |
| [133] |
R.M. Palmer, A.G. Ferrige, S. Moncada, Nature 327 (1987) 524-526. DOI:10.1038/327524a0 |
| [134] |
D. Basudhar, L.A. Ridnour, R. Cheng, et al., Coord. Chem. Rev. 306 (2016) 708-723. DOI:10.1016/j.ccr.2015.06.001 |
| [135] |
D. Fukumura, S. Kashiwagi, R.K. Jain, Nat. Rev. Cancer 6 (2006) 521-534. DOI:10.1038/nrc1910 |
| [136] |
A.J. Burke, F.J. Sullivan, F.J. Giles, S.A. Glynn, Carcinogenesis 34 (2013) 503-512. DOI:10.1093/carcin/bgt034 |
| [137] |
V. Somasundaram, R. Nadhan, K.H. S, et al., Crit. Rev. Oncol. Hematol. 101 (2016) 184-192. DOI:10.1016/j.critrevonc.2016.03.004 |
| [138] |
Z. Huang, J. Fu, Y. Zhang, J. Med. Chem. 60 (2017) 7617-7635. DOI:10.1021/acs.jmedchem.6b01672 |
| [139] |
S. Mocellin, V. Bronte, D. Nitti, Med. Res. Rev. 27 (2007) 317-352. DOI:10.1002/med.20092 |
| [140] |
M.M. Reynolds, S.D. Witzeling, V.B. Damodaran, et al., Biochem. Biophys. Res. Commun. 431 (2013) 647-651. DOI:10.1016/j.bbrc.2013.01.041 |
| [141] |
H. Vahora, M.A. Khan, U. Alalami, A. Hussain, J. Cancer Prev. 21 (2016) 1-12. DOI:10.15430/JCP.2016.21.1.1 |
| [142] |
A. Kamm, P. Przychodzen, A. Kuban-Jankowska, et al., Nitric Oxide 93 (2019) 102-114. DOI:10.1016/j.niox.2019.09.005 |
| [143] |
J. Scicinski, B. Oronsky, S. Ning, et al., Redox Biol. 6 (2015) 1-8. DOI:10.1016/j.redox.2015.07.002 |
| [144] |
C.A. Carter, B. Oronsky, S. Caroen, et al., Respir. Med. Case Rep. 18 (2016) 62-65. |
| [145] |
L.K. Keefer, ACS Chem. Biol. 6 (2011) 1147-1155. DOI:10.1021/cb200274r |
| [146] |
J.A. Hrabie, L.K. Keefer, Chem. Rev. 102 (2002) 1135-1154. DOI:10.1021/cr000028t |
| [147] |
J.E. Saavedra, A. Srinivasan, C.L. Bonifant, et al., J. Org. Chem. 66 (2001) 3090-3098. DOI:10.1021/jo0016529 |
| [148] |
C. Chen, Y. Shi, S. Li, et al., Arch. Pharm. (Weinheim) 339 (2006) 366-371. DOI:10.1002/ardp.200500262 |
| [149] |
N. Barraud, B.G. Kardak, N.R. Yepuri, et al., Angew. Chem. Int. Ed. 51 (2012) 9057-9060. DOI:10.1002/anie.201202414 |
| [150] |
K. Sharma, A. Iyer, K. Sengupta, H. Chakrapani, Org. Lett. 15 (2013) 2636-2639. DOI:10.1021/ol400884v |
| [151] |
Y. Zou, C. Yan, E.E. Knaus, et al., RSC Adv. 7 (2017) 18893-18899. DOI:10.1039/C7RA00401J |
| [152] |
J. Hou, Y. Pan, D. Zhu, et al., Nat. Chem. Biol. 15 (2019) 151-160. DOI:10.1038/s41589-018-0190-5 |
| [153] |
M.L. O'Brien, K.D. Tew, Eur. J. Cancer 32a (1996) 967-978.
|
| [154] |
S. Mahajan, W.M. Atkins, Cell. Mol. Life Sci. 62 (2005) 1221-1233. DOI:10.1007/s00018-005-4524-6 |
| [155] |
Z. Huang, J. Wu, Y. Zou, et al., J. Med. Chem. 61 (2018) 1833-1844. DOI:10.1021/acs.jmedchem.7b01178 |
| [156] |
R. Xue, J. Wu, X. Luo, et al., Org. Lett. 18 (2016) 5196-5199. DOI:10.1021/acs.orglett.6b02222 |
| [157] |
X. Luo, J. Wu, T. Lv, et al., Org. Chem. Front. 4 (2017) 2445-2449. DOI:10.1039/C7QO00695K |
| [158] |
T. Lv, J. Wu, F. Kang, et al., Org. Lett. 20 (2018) 2164-2167. DOI:10.1021/acs.orglett.8b00423 |
| [159] |
J.F. Quinn, M.R. Whittaker, T.P. Davis, J. Control. Release 205 (2015) 190-205. DOI:10.1016/j.jconrel.2015.02.007 |
| [160] |
A.C. Midgley, Y. Wei, Z. Li, et al., Adv. Mater. 32 (2019) e1805818. |
| [161] |
H. Alimoradi, K. Greish, A.B. Gamble, G.I. Giles, Pharm. Nanotechnol. 7 (2019) 279-303. DOI:10.2174/2211738507666190429111306 |
| [162] |
X. Jia, Y. Zhang, Y. Zou, et al., Adv. Mater. 30 (2018) e1704490. DOI:10.1002/adma.201704490 |
| [163] |
Y. Hu, T. Lv, Y. Ma, et al., Nano Lett. 19 (2019) 2731-2738. DOI:10.1021/acs.nanolett.9b01093 |
| [164] |
E.L. Sievers, P.D. Senter, Annu. Rev. Med. 64 (2013) 15-29. DOI:10.1146/annurev-med-050311-201823 |
| [165] |
R.V. Chari, M.L. Miller, W.C. Widdison, Angew. Chem. Int. Ed. 53 (2014) 3796-3827. |
| [166] |
F. Sun, Y. Wang, X. Luo, et al., Cancer Res. 79 (2019) 3395-3405. |
| [167] |
R. Kumar, Curr. Med. Chem. 23 (2016) 2637-2642. DOI:10.2174/0929867323666160812150737 |
| [168] |
X. Zhan, Y. Huang, S. Qian, Curr. Med. Chem. 25 (2018) 3435-3454. DOI:10.2174/0929867325666180221140745 |
| [169] |
M.C. Franco, K.C. Ricart, A.S. Gonzalez, et al., J. Biol. Chem. 290 (2015) 19055-19066. DOI:10.1074/jbc.M115.663278 |
| [170] |
S.N. Topkaya, V.H. Ozyurt, A.E. Cetin, S. Otles, Biosens. Bioelectron. 102 (2018) 464-469. DOI:10.1016/j.bios.2017.11.061 |
| [171] |
V. Fernando, X. Zheng, Y. Walia, et al., Antioxidants 8 (2019) 404-435. DOI:10.3390/antiox8090404 |
| [172] |
M.M. Lerch, M.J. Hansen, G.M. van Dam, et al., Angew. Chem. Int. Ed. 55 (2016) 10978-10999. DOI:10.1002/anie.201601931 |
| [173] |
V. San Miguel, C.G. Bochet, A. del Campo, J. Am. Chem. Soc. 133 (2011) 5380-5388. DOI:10.1021/ja110572j |
| [174] |
K. Hull, J. Morstein, D. Trauner, Chem. Rev. 118 (2018) 10710-10747. DOI:10.1021/acs.chemrev.8b00037 |
| [175] |
M.J. Hansen, W.A. Velema, M.M. Lerch, et al., Chem. Soc. Rev. 44 (2015) 3358-3377. DOI:10.1039/C5CS00118H |
| [176] |
W.A. Velema, W. Szymanski, B.L. Feringa, J. Am. Chem. Soc. 136 (2014) 2178-2191. DOI:10.1021/ja413063e |
| [177] |
R. Horbert, B. Pinchuk, P. Davies, et al., ACS Chem. Biol. 10 (2015) 2099-2107. DOI:10.1021/acschembio.5b00174 |
| [178] |
M.J. Hansen, F.M. Feringa, P. Kobauri, et al., J. Am. Chem. Soc. 140 (2018) 13136-13141. DOI:10.1021/jacs.8b04870 |
| [179] |
Z. Li, K. Su, Z. Jiang, et al., J. Med. Chem. 62 (2019) 7583-7588. DOI:10.1021/acs.jmedchem.9b00688 |
| [180] |
G. Bassolino, C. Nancoz, Z. Thiel, et al., Chem. Sci. 9 (2018) 387-391. DOI:10.1039/C7SC03627B |
| [181] |
R. Tamura, A. Balabanova, S.A. Frakes, et al., J. Med. Chem. 62 (2019) 1959-1970. DOI:10.1021/acs.jmedchem.8b01508 |
| [182] |
A.S. Lubbe, W. Szymanski, B.L. Feringa, Chem. Soc. Rev. 46 (2017) 1052-1079. DOI:10.1039/C6CS00461J |
| [183] |
D. Lemoine, C. Habermacher, A. Martz, et al., Proc. Natl. Acad. Sci. U. S. A. 110 (2013) 20813-20818. DOI:10.1073/pnas.1318715110 |
| [184] |
N.J. Hauwert, T.A.M. Mocking, D. Da Costa Pereira, et al., J. Am. Chem. Soc. 140 (2018) 4232-4243. DOI:10.1021/jacs.7b11422 |
| [185] |
X. Gómez-Santacana, S.M. de Munnik, P. Vijayachandran, et al., Angew. Chem. Int. Ed. 57 (2018) 11608-11612. DOI:10.1002/anie.201804875 |