 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Magnetic Resonance and Atomic and Molecular Physics, National Center for Magnetic Resonance in Wuhan, Wuhan Institute of Physics and Mathematics, Chinese Academy of Sciences, Wuhan 430071, China;
c School of Mechanical & Electronic Engineering, Nanchang University, Nanchang 330031, China
Photocatalytic hydrogen evolution from water is one of the most promising green technology for direct conversion of solar energy to chemical energy and energy storage. In the past few decades, it has attracted immense efforts for developing various photocatalysts to produce H2 through solar water splitting [1-4]. Recently, graphitic carbon nitride (g-C3N4), a metal-free photocatalyst, has received extensive attention due to its good photocatalytic hydrogen and oxygen production of water splitting [5-7], water oxidation [8, 9], photocatalytic degradation of organic pollutants [10-12], organic reaction catalyst [13, 14], catalyst carrier and energy storage material [15-17].
Currently, tremendous efforts have been focused on reporting synthesis of g-C3N4 by thermal treatment of different nitrogen-rich precursors under different reaction conditions [18-20]. For example, Martin research group reported that g-C3N4 can be prepared by using different precursors such as urea, melamine or dicyandiamide under same high-temperature calcination and ambient atmosphere conditions [21-26]. Subsequent reports on large amounts of urea-derived g-C3N4 outperformed the thiourea and dicyandiamide-derived counterparts in the photocatalytic hydrogen evolution from water splitting [27, 28]. Large numbers of refreshing literatures have reported on the effect of calcination temperature and precursor treatment on the synthesis, structure and properties of the g-C3N4 [29, 30]. These reports indicated that different precursor and its reaction conditions of high-temperature calcination, ambient atmosphere and special precursor treatment methods can affect the physicochemical properties of g-C3N4, such as specific surface area, light absorption edge and nanostructures [31-33]. However, these literatures did not involve the influence of the preservation time and morphology of the same precursor on the thermal polymerization method, structure and photocatalytic property of g-C3N4.
In addition, many modification strategies have been reported to improve the photocatalytic performance of g-C3N4, including alkali-assisted synthesis, phosphorous-mediated synthesis and acid pretreatment of the precursors [34-36]. These strategies can generate functional groups (such as -OH, -COOH, -C≡N and -CONH2) via defects formation to increase active sites, enhance light absorption and change the electronic band structure of g-C3N4. For example, -C≡N can be formed in the g-C3N4 by alkali-assisted synthesis to improve the photocatalytic performance of g-C3N4 [37]. In addition, -CONH2 can be formed in the g-C3N4 by acid pretreatment to improve the photocatalytic performance of g-C3N4 [38]. However, these strategies are not environmental-friendly, complex and expensive because they need supply additives or special auxiliary conditions during the synthetic process and result in environmental pollution, such as usage of strong acid and formation of H2S. Therefore, it is urgent to develop a facile and environmental friendly approach for in-situ formation functional groups or defects in the g-C3N4 via direct calcination of N-rich precursors, thereby improve the photocatalytic performance of g-C3N4.
Herein, we tried to synthesize g-C3N4 with functional groups by calcination of the same precursorwith different kept-time. When the fresh urea and the urea kept for five years were used as precursors, it can obtain g-C3N4 with different color and photocatalytic activity. The g-C3N4 obtained from the fresh urea and the urea kept for five years are yellow and brown, respectively, and they are hereafter named as Y-GCN and C-GCN, respectively. Noteworthily, C-GCN exhibits more than 5 times higher photocatalytic H2 generation rate than that of Y-GCN. We systematically investigated the structures, morphologies, and the hydrogen evolution properties of Y-GCN and C-GCN. Experimental results show that a new kind of functional group of -N=CH- was firstly introduced into the C-GCN, resulting in enhancement of visible light absorption and variation of band structure and improvement of charge carrier separation. The present work not only provides a new idea to prepare g-C3N4 with special defects but also demonstrates the introduction of -N=CH- groups into g-C3N4 is an effective approach of "defect engineering" to improve the photocatalytic performance of g-C3N4 for the first time.
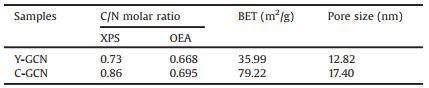
The photocatalytic hydrogen evolution of C-GCN and Y-GCN were evaluated under irradiation of visible light using triethanol-amine as holes scavenger. As shown in Fig. 1a, in the absence of Pt co catalysts, the H2 evolution rate of C-GCN is 26.5 μmol g-1 h-1, which is about 10 times of Y-GCN (2.8 μmol g-1 h-1). After loaded with Pt cocatalysts via in situ photo reduction of H2PtCl6, the H2 evolution rates of C-GCN and Y-GCN are both greatly improved due to platinum's high conductivity, negligible over potential for H2 evolution and excellent electron transport kinetic. As shown in Fig. 1b, the hydrogen evolution rate of C-GCN and Y-GCN is 1953 and 361.5 μmol g-1 h-1, respectively, when Pt was loaded on the surface of catalysts. The H2 evolution rates of C-GCN and Y-GCN nearly become nearly unchanged over 20 h photoreaction, indicating that they show good photo stability (Fig. 1c). In terms of apparent quantum efficiency (AQE) at 420 nm, C-GCN has a value of 6.3% and nearly 7.3 times higher than that of Y-GCN at 0.86%.

|
Download:
|
| Fig. 1. (a) Hydrogen evolution rate to Y-GCN and C-GCN without Pt in 80 mL of aqueous solution under visible-light irradiation. (b) Pt-loaded Y-GCN and C-GCN photocatalysts in aqueous solution under visible-light irradiation. (c) Time course of hydrogen evolution over 20 h for C-GCNand Y-GCN. | |
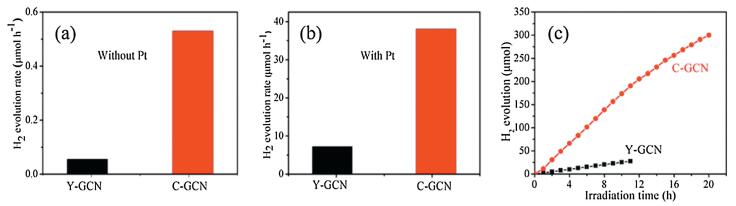
To explore the marked difference of the photocatalytic performance between C-GCN and Y-GCN, some tests were used. As shown in Fig. 2a, the C-GCN powder is gray, while Y-GCN is faint yellow. Compared with Y-GCN, C-GCN has much stronger absorption intensity in the visible light region and the absorption edge obviously red-shifts. As shown in Fig. 2b, the calculated bandgaps of Y-GCN and C-GCN are 2.78 eV and 2.66 eV, respec-tively. As shown in Fig. 2c, C-GCN exhibits much better photocurrent than Y-GCN. C-GCN shows lower intensity of PL spectra than Y-GCN (Fig. 2d). The above results indicate that C-GCN has much better light absorption and more efficient separation rate of photo generated electrons and holes than Y-GCN.

|
Download:
|
| Fig. 2. (a) DRS and (b) plots of (ahv)2 vs. hv over the Y-GCN and C-GCN (Inset in (a) shows a digital photograph of Y-GCN and C-GCN). (c) Transient photocurrent response of Y-GCN and C-GCN under visible-light illumination. (d) Photoluminescence spectra of Y-GCN and C-GCN at an excitation wavelength of 375 nm. | |
The crystal and molecular structure of Y-GCN and C-GCN were systematically researched in order to make clear their different photocatalytic performance. Fig. 3 shows the results of 13C and 15N MAS NMR of Y-GCN and C-GCN. The 13C spectra of two samples show two resonance peaks at δ1 165 ppm and δ2 156.7 ppm (Fig. 3a). The peak at 165.4 ppm is ascribed to the C(2) atoms in CN2, while the peak at 156.7 ppm is assigned to C(1) atoms in CN3 groups [39, 40]. The results are well consistent with those reported by Wang et al., confirming the C-GCN possesses the g-C3N4 framework. In addition, a new signal at 163.4 ppm (C3) appears in the C-GCN, which can be ascribed to the imine signal, indicating that the peripheral -N=CH- groups exist in C-GCN [41]. Four peaks in the 15N spectra of Y-GCN and C-GCN can be assigned (Fig. 3b). The broad signal at the –199.9 ppm corresponds to the peripheral nitrogens (N1) in the heptazine ring; the weak peak at –235.1 ppm is assigned to the center nitrogen (N2) of heptazine; the strongest peak at –254.2 ppm is attributed to the secondary amine (N3: -NH group); and the 15N signal at –274 ppm is ascribed to amine (N4: -NH2 group) [42, 43]. The above four peaks coincide with those of g-C3N4, indicating the g-C3N4 structure exists in C-GCN and Y-GCN. In addition, a new signal at –285.4 ppm (N5) among the C-GCN coincides with the urea NH2 signal, demonstrating a new peripheral amine groups were formed in C-GCN [38]. Compared to the structure of Y-GCN (Fig. 3c), the NMR results confirm the existence of peripheral -N=CH- NH2 groups in C-GCN (Fig. 3d).

|
Download:
|
| Fig. 3. (a) 13C and (b) 15N solid state NMR spectra of Y-GCN and C-GCN. (c) The molecular structure of Y-GCN and (d) C-GCN. (e) Network structure of C-GCN. | |
In addition, carbon and nitrogen contents of Y-GCN and C-GCN samples were determined by elemental analyses. As shown in Table 1, the C/N atomic ratio of Y-GCN and C-GCN is 0.668 and 0.695, respectively, lower than the theoretical value of 0.75 for g-C3N4, which could be due to the existence of many amino groups (C-NHx) [27]. The larger C/N atomic ratio of C-GCN than that of Y-GCN also indicates the existence of peripheral -N=CH- group in the C-GCN. Furthermore, as shown in Table 1, the organic elemental analysis (OEA) shows the C/N atomic ratio of 0.695 for C-GCN is consistent with the calculated C/N atomic ratio (0.7) of the polymeric structure for the -N=CH- NH2 group modified g-C3N4 (Fig. 3e). The result is consistent with the above analysis of NMR results.
|
|
Table 1 The physicochemical properties of Y-GCN and C-GCN. |
{kind=link}
{kind=link}
{kind=link}
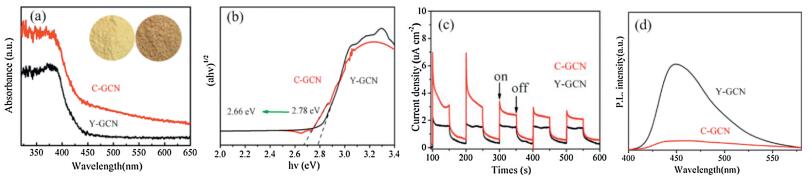
As shown in Fig. 4a, the XRD patterns of C-GCN are similar to that of Y-GCN. Two obvious featured peaks are found in Y-GCN and C-GCN. The weak peak at 12.9°, indexed as the (100) of g-C3N4, confirms that the tri-s-triazine-based structure is retained in C-GCN [19]. The strong peak at 27.8° is indexed as (002) of g-C3N4, which is ascribed to the interlayer stacking of conjugated aromatic rings [44]. Noteworthily, the (002) peak shifts from 27.8 of Y-GCN to 27.4 of C-GCN, suggesting that the crystal structure of the C-GCN tends to become less stable than Y-GCN [45]. Furthermore, there are two other weak peaks at 17.6 and 21.4 in the C-GCN, corresponding to (011) and (110) of g-C3N4 [46]. The results confirm that both Y-GCN and C-GCN show the structure of g-C3N4.

|
Download:
|
| Fig. 4. XRD patterns (a) and FT-IR spectra (b) of Y-GCN and C-GCN. High-resolution XPS of spectra of C 1s (c) and N 1s (d) of Y-GCN and C-GCN. | |
{kind=link}
As shown in Fig. 4b, FT-IR spectrum of C-GCN is similar to that of Y-GCN. Among them, there are five characteristic peaks at 810, 1240, 1320, 1411, 1459, and 1638 cm-1, which are attributed to the skeletal vibrations of tri-s-triazine ring units of g-C3N4 [47, 48]. The peaks at 3170 cm-1 and 3277 cm-1 are ascribed to the stretching vibration of –NH2 and -OH, respectively [49, 50]. And the peak at 1565 cm-1 is assigned to the stretching vibration of N=C group [51, 52]. Compared with the Y-GCN, the peak's intensity of N=C group markedly increase among the C-GCN. The above results indicate that the units of tri-s-triazine ring exist in the C-GCN and Y-GCN, and there could exist peripheral -N=C- group in the C-GCN.
The wide-scan XPS spectra of Y-GCN and C-GCN confirm the existence of C, N, and O elements in Y-GCN and C-GCN (Fig. S1 in Supporting information). As shown in Fig. 4c, there are three peaks at 288.26, 285.96 and 284.72 eV among Y-GCN and C-GCN, corresponding to N-C=N in the framework of g-C3N4, C-NHx (x = 1 and 2) on the edges of heptazine units and adventitious hydrocarbons (C-C/C=C), respectively [53]. Noteworthily, com-pared with Y-GCN, C-NHx signal exhibits a small shift to higher binding energy in C-GCN, which can be taken as good evidence for the formation of new defect group in C-GCN [54]. Furthermore, there is additional peak at 284.78 eV among the C1s spectra of C-GCN that is attributed to –C–H species, implying a new group of –C–H in C-GCN [55]. As shown in Fig. 4d, the N1 s spectra of Y-GCN and C-GCN can be deconvoluted into three peaks at 398.74, 399.76 and 401.2 eV, which can be ascribed to bicoordinated (N2C) and tri-coordinated (N3C) nitrogen atoms and C-NHx (x = 1 and 2) groups in the heptazine framework, respectively [56, 57]. Compared with Y-GCN, the peaks of N2C among C-GCN slightly shift to higher binding energy, which could be due to the generation of terminal CH=N groups whose N 1s binding energy are intermediate between those of N2C and N3C in C-GCN (Table S1 in Supporting information) [58]. These results are consistent with those reported in the literature. Furthermore, the elementary analysis based on XPS data shows that the C/N ratio increases from 0.73 in Y-GCN to 0.86 in the C-GCN (Table 1), confirming the existence of terminal group of CH=N in C-GCN. Therefore, based on the NMR, FTIR and XPS results, it can be concluded that the peripheral -N=CH- NH2 groups appear in C-GCN but not in Y-GCN.
The morphologies of Y-GCN and C-GCN were investigated (Fig. 5). The Y-GCN and C-GCN consist of large amounts of packing layers with different sizes of nanosheets (Figs. 5a and b). As shown in Figs. 5c and d, C-GCN shows more highly open-up flat and more ultrathin layered structure than Y-GCN, resulting that the surface area and pore volume of C-GCN are larger than that of the Y-GCN. As shown in Fig. S2 (Supporting information) and Table 1, the surface areas of the Y-GCN and C-GCN are 35.99 m2/g and 79.22 m2/g, respectively.

|
Download:
|
| Fig. 5. SEM images of (a) Y-GCN and (b) C-GCN. TEM images of (c) Y-GCN and (d) C-GCN. | |
{kind=link}
Thus, the results of SEM, TEM, BET, IR, XPS and NMR indicate that the calcination of urea with different kept-time can prepare g-C3N4 with different morphology, surface areas, polymerization's types, and molecular structure, which further lead to different photocatalytic performance of H2 evolution. It should be noted that this is first report on polymeric carbon nitride frameworks highly relying on its precursors with different kept-time.
To explain why the reactant of urea with different kept-time lead to different structures, morphologies and photocatalytic performances of g-C3N4, we further investigated the precursor's nature. As shown in Fig. S3a and Table S2 (Supporting information), the results of XRD and elemental analyses show that there are no obvious difference of crystal and structure between urea-1 (the fresh urea) and urea-2 (urea kept for five years). Fig. S3b (Supporting information) shows that some differences appeared in thermal weight analysis of the two kind of precursor. There is obvious particular process among the calcination process of the urea-2 at T = 230–243 ℃ (process (1)) that is not exist in the calcination process of urea-1, which is ascribed to the interaction among biuret and cyanuric acid, indicating the synthetic process route of C-GCN using the urea-2 is different from that of Y-GCN using the urea-1 among thermal treatment process. The results confirm that kept-time of urea can affect internal polymerization process of g-C3N4. Fig. S4 (Supporting information) shows that urea-2 is conglomerate and branched, while urea-1 possesses smooth rod structure, indicating different kept-time of urea make significant effect on the morphology of urea, further lead to difference in the structure and optical properties of the as-prepared g-C3N4. Therefore, higher photocatalytic performance of C-GCN than Y-GCN could be ascribed that differences of precursor's morphology lead to differences in specific synthetic pathways.
In order to understand the effect of terminal defects on g-C3N4, UV–vis diffuse reflection spectra (DRS) and Mott-Schottky plots of Y-GCN and C-GCN were investigated. As shown in Figs. 2a and b, the DRS spectra of the as prepared Y-GCN and C-GCN show that the calculated bandgaps are 2.78 and 2.66 eV for the Y-GCN and C-GCN, respectively. Figs. S5a and b (Supporting information) show the Mott-Sckottky plots of Y-GCN and C-GCN being working electrode (0.5 mol/L K2SO4 as electrolyte), respectively. The flat band potential (Vfb) as calculated from the x intercepts of the linear region are found to be –0.78 and –0.62 eV vs. SCE (equivalent to 0.54 and 0.38 eV vs. NHE, respectively) for Y-GCN and C-GCN, respectively. It is known that the flat band potential of n-type semiconductor equals to the Fermi level (EF). Therefore, the EF values of Y-GCN and C-GCN are 0.54 and 0.38 eV, respectively. Accordingly, the CB of Y-GCN and C-GCN can be calculated as –0.74 and –0.58 eV, respectively. The band positions can be calculated by the following empirical formulas: ECB= EVB – Eg, and the valence band (VB) of Y-GCN and C-GCN are 2.04 and 2.08 eV, respectively (Fig. S6 in Supporting information) [59, 60]. These data suggest that the introduction of terminal -N=CH- groups into g-C3N4 among the C-GCN can affect the position of CB of the C-GCN, leading to the enhancement of photocatalytic performance of H2 generation.
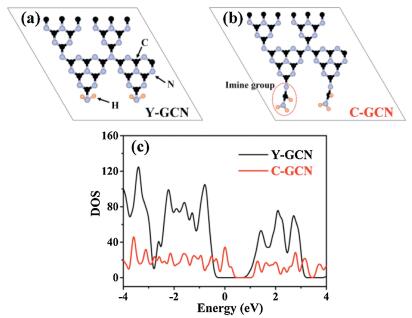
In order to well understand the effect of the terminal -N=CH- groups on the band structure of C-GCN, density of states (DOS) and partial density of states (PDOS) of the Y-GCN and C-GCN were performed (Fig. 6 and Fig. S6). As shown in Figs. S6a and b, both C 2p and N 2p orbitals contribute to the CB of Y-GCN, while the VB is mainly composed of C 2s and C 2p orbitals, in consistent with the previously reported results [61]. As shown in Figs. S6c and d, the CB of C-GCN is also composed of both C 2p and N 2p orbitals, whereas the VB of C-GCN is ascribed to C 2s, C 2p and N 2p orbitals. As the band alignments shown in Fig. S7 (Supporting information), the DOS shows that after introducing imine groups (-N=CH-) into the unit cell of g-C3N4, it narrows the band gap of g-C3N4 to enhance its visible light absorption, which agree well with the above results of the DRS and Mott-Sckottky plots. The results confirmed that the introduction of terminal -N=CH- groups can affect electron density, CB and VB of g-C3N4 in C-GCN. The analyses also confirm the difference of molecular structure, band structure and photocatalytic performance in C-GCN and Y-GCN.

|
Download:
|
| Fig. 6. Structural models of (a) Y-GCN and (b) C-GCN, respectively. (c) DOS of Y-GCN and C-GCN. | |
{kind=link}
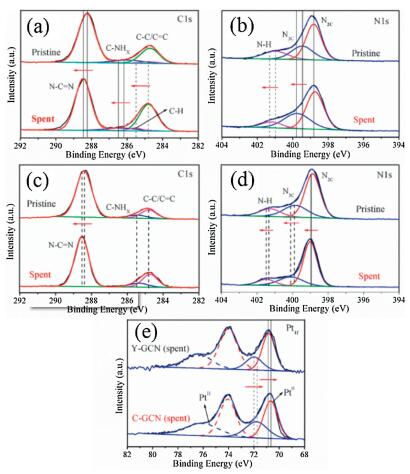
Besides the above-discussed influence of functional groups of -N=CH- on the photocatalytic performance of H2 production of g-C3N4, the factor of cocatalyst of Pt was systematically investigated. In the presence of Pt cocatalyst, the rate of H2 production of the C-GCN is much larger than that of Y-GCN. The marked difference in photocatalytic efficiency between Y-GCN and C-GCN could arise from different transfer rates from the internal sites of the catalysts to the surface of Pt particles, depending on the coupling between active sites (N=CH) and Pt particles. To further explore interaction of C-GCN with Pt cocatalyst, the catalyst with in-situ photo deposited Pt after 20 h photocatalytic reaction was characterized by XPS and XRD. As shown in Fig. S8 (Supporting information), XRD shows the spent C-GCN has two characteristic peaks of g-C3N4 at 13.1° and 27.5°, while the 39.8° and 46.5° signals can be attributed to the presence of the Pt cocatalyst. The results confirm that Pt particles are successfully deposited on the surface of C-GCN, and C-GCN is very stable during the photocatalytic reaction. As shown in Figs. 7a and b, compared to the pristine C-GCN, the C1 sspectra of the spent C-GCN show three signals shifted to higher bind energy (B.E.) by over 0.3 eV, 0.3 eV and 0.7 eV for the N C=N, C-NHx and C-H signals, respectively. The N1 sspectra of the spent C-GCN show two signals shifted to higher B.E. by over 0.4 and 0.3 eV for the N3C and N-H signals, respectively. In addition, as shown in Figs. 7c and d, compared to the pristine Y-GCN, the C1sspectra of the spent Y-GCN shows N–C=N signal shifted to higher B.E. by over 0.15 eV, while N 1s spectra of the spent Y-GCN show two signals shifted to higher B.E. by over 0.2 eV and 0.1 eV for the N3C and N-H signals, respectively. After 20 h photocatalytic reaction, the B.E. value of C and N in Y-GCN and C-GCN all became larger than the pristine one, whereas there are larger shifts in the C-GCN than Y-GCN. The results indicate the electron density of C-GCN become lower than that of Y-GCN. Furthermore, as shown in Fig. 7e, the B.E. of Pt0 on the spent C-GCN is 70.6 eV, smaller than that on the spent Y-GCN (70.9 eV). The B.E. of PtⅡ on C-GCN (71.8 eV) is smaller than that on the spent Y-GCN (72.0 eV), which can be assigned to Pt2+ of PtO [62]. The directions of the shifts toward higher B.E. for the carbon and nitride and lower B.E. for Pt indicate the donation of electron density from the carbon and nitride of Y-GCN and C-GCN to Pt cocatalyst, in accord with metal-support interaction reported in the literatures[63]. But the larger shifts occurred in C-GCN than in Y-GCN, suggesting C-GCN has stronger interaction than Y-GCN with Pt cocatalyst, resulting in facilitating charge transfer and significant increase in photocatalytic hydrogen evolution. These XPS results demonstrate that the existence of -N=CH- groups in C-GCN leads to tight interaction between C-GCN and Pt nanoparticles, and then improves the charge separation and photocatalytic performance.

|
Download:
|
| Fig. 7. (a) XPS for C 1s spectrum and (b) N 1s spectrum of the pristine C-GCN and that with in-situ photodeposited Pt after 20 h photocatalytic reaction. (c) XPS for C 1s spectrum and (d) N1s spectrum of the pristine Y-GCN and that with in-situ photodeposited Pt after 20 h photocatalytic reaction. (e) XPS of Pt on the surface of the C-GCN and Y-GCN with in-situ photodeposited Pt after 20 h photocatalytic reaction. | |
{kind=link}
On the basis of the above experimental results, we proposed the mechanism of the photocatalytic hydrogen production of water splitting over the as-prepared catalysts. As shown in Scheme 1, under irradiation of visible light, C-GCN can directly absorb light of 420–520 nm to generate electrons (e-) and holes (h+) and the electrons are excited from VB to CB. Firstly, the photo generated electrons on the CB of the C-GCN can quickly transfer to Pt nanoparticles because there are tight interactions between C-GCN and Pt nanoparticles via -N=CH- groups. Meanwhile, the holes on the VB of C-GCN are reacted with the sacrificial agent of triethanolamine to finish the oxidation process, which improves the separation of photo generated electrons and holes. Therefore, there is effective charge separation and the separated electrons have enough time to reduce H2O to H2 on the surface of C-GCN. As for Y-GCN, the photocatlytic mechanism is similar to that of C-GCN. But the photo-generated electrons on the CB of Y-GCN have smaller transfer rate than those on the CB of C-GCN to Pt nanoparticles because there exist functional groups of -N=CH- in C-GCN but not in Y-GCN. The mechanism explains why C-GCN exhibits much better activity for hydrogen evolution than Y-GCN.

|
Download:
|
| Scheme 1. Proposed photocatalytic mechanism for hydrogen evolution over C-GCN and Y-GCN under visible light irradiation. | |
{kind=link}
In this paper, Y-GCN and C-GCN were firstly prepared by using the fresh urea and the urea kept for five years as precursors, respectively. The photocatalytic hydrogen evolution rate of C-GCN is much larger 5 times than that of Y-GCN. Different from Y-GCN, results of XPS, NMR, DRS indicate that there are functional groups of -N=CH- introduced in the C-GCN. Noteworthily, the introduction of functional groups of -N=CH- result in enhancement of visible light absorption, high charge-separation efficiency, suitable band structure, and strong interaction between C-GCN and Pt, and then lead to much improvement of photocatalytic activity. DFT calculation also confirms the introduction of -N=CH- can change the band structure of g-C3N4. The present work provides a new strategy to improve the photocatalytic performance of g-C3N4 via the introduction of functional groups of N=CH.
AcknowledgmentsWe gratefully acknowledge the financial support of the National Natural Science Foundation of China (NSFC, Nos. 51622806, 51878325, 51868050, 51378246 and 51720105001) and the Natural Science Foundation of Jiangxi Province (Nos. 20162BCB22017, 20165BCB18008, 20171ACB20017, 20133ACB21001 and 20171BAB206049).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.020.
| [1] |
A. Fujishima, K. Honda, Nature 238 (1972) 37-38. DOI:10.1038/238037a0 |
| [2] |
A. Xie, Z.H. Pan, M. Yu, et al., Chin. Chem. Lett. 30 (2019) 225-228. |
| [3] |
A. Kudo, Y. Miseki, Chem. Soc. Rev. 38 (2009) 253-278. DOI:10.1039/B800489G |
| [4] |
Z. Han, F. Qiu, R. Eisenberg, P.L. Holland, T.D. Krauss, Science 338 (2012) 1321-1324. DOI:10.1126/science.1227775 |
| [5] |
Y.C. Nie, F. Yu, L.C. Wang, et al., Appl. Catal. B:Environ. 227 (2018) 312-321. DOI:10.1016/j.apcatb.2018.01.033 |
| [6] |
Y. Wang, X. Wang, M. Antonietti, Angew. Chem. Int. Ed. 51 (2012) 68-89. DOI:10.1002/anie.201101182 |
| [7] |
J. Wang, J. Chen, P. Wang, J. Hou, C. Wang, Y. Ao, Appl. Catal. B:Environ. 239 (2018) 578-585. DOI:10.1016/j.apcatb.2018.08.048 |
| [8] |
Y. Hou, A.B. Laursen, J. Zhang, et al., Angew. Chem. Int. Ed. 52 (2013) 3621-3625. DOI:10.1002/anie.201210294 |
| [9] |
C.C. Wang, X.H. Yi, P. Wang, Appl. Catal. B:Environ. 247 (2019) 24-48. |
| [10] |
S.Z. Wu, C.H. Chen, W.D. Zhang, Chin. Chem. Lett. 25 (2014) 1247-1251. DOI:10.1016/j.cclet.2014.05.017 |
| [11] |
X.H. Jiang, Q.J. Xing, X.B. Luo, et al., Appl. Catal. B:Environ. 228 (2018) 29-38. DOI:10.1016/j.apcatb.2018.01.062 |
| [12] |
X.H. Jiang, L.C. Wang, F. Yu, J.P. Zou, et al., ACS Sustain. Chem. Eng. 6 (2018) 12695-12705. DOI:10.1021/acssuschemeng.8b01695 |
| [13] |
L. Jiang, X. Yuan, G. Zeng, et al., Environ. Sci. Nano 5 (2018) 599-615. |
| [14] |
H. She, Y. Sun, L. Wang, et al., Appl. Catal. B:Environ. 245 (2019) 439-447. DOI:10.1016/j.apcatb.2019.01.002 |
| [15] |
Y.P. Wang, Y.K. Li, J.L. Zhao, et al., Int. J. Hydrogen Energy 44 (2019) 618-628. DOI:10.1016/j.ijhydene.2018.11.067 |
| [16] |
J.P. Zou, D.D. Wu, J. Luo, et al., ACS Catal. 6 (2016) 6861-6867. DOI:10.1021/acscatal.6b01729 |
| [17] |
J.P. Zou, Y. Chen, S. S.-Liu, et al., Water Res. 150 (2019) 330-339. |
| [18] |
K. Maeda, X. Wang, Y. Nishihara, et al., J. Phys. Chem. C 113 (2009) 4940-4947. |
| [19] |
X. Wang, K. Maeda, A. Thomas, et al., Nat. Mater. 8 (2009) 76-80. DOI:10.1038/nmat2317 |
| [20] |
J. Low, S. Cao, J. Yu, S. Wageh, Chem. Commun. 50 (2014) 10768-10777. DOI:10.1039/C4CC02553A |
| [21] |
Q. Su, J. Sun, J. Wang, et al., Catal. Sci. Technol. 4 (2014) 1556-1562. DOI:10.1039/c3cy00921a |
| [22] |
M. Ding, J.J. Zhou, H.C. Yang, et al., Chin. Chem. Lett. 31 (2020) 71-76. DOI:10.1016/j.cclet.2019.05.029 |
| [23] |
Y. Zhang, H. Zhang, L. Cheng, et al., RSC Adv. 6 (2016) 14002-14008. DOI:10.1039/C5RA22732A |
| [24] |
Z. Zhou, J. Wang, J. Yu, et al., J. Am. Chem. Soc. 137 (2015) 2179-2182. DOI:10.1021/ja512179x |
| [25] |
H.S. Zhai, L. Cao, X.H. Xia, Chin. Chem. Lett. 24 (2013) 103-106. DOI:10.1016/j.cclet.2013.01.030 |
| [26] |
Q. Liu, D.B. Zhu, M.L. Guo, et al., Chin. Chem. Lett. 30 (2019) 1639-1642. DOI:10.1016/j.cclet.2019.05.058 |
| [27] |
D.J. Martin, K. Qiu, S.A. Shevlin, et al., Angew. Chem. Int. Ed. 53 (2014) 9240-9245. DOI:10.1002/anie.201403375 |
| [28] |
Y.S. Jun, J. Park, S.U. Lee, et al., Angew. Chem. Int. Ed. 125 (2013) 11289-11293. DOI:10.1002/ange.201304034 |
| [29] |
F. Dong, Z. Wang, H. Zhang, et al., J. Colloid. Interf. Sci. 401 (2013) 70-79. DOI:10.1016/j.jcis.2013.03.034 |
| [30] |
J. Xu, L. Zhang, R. Shi, Y. Zhu, J. Mater. Chem. A 1 (2013) 14766-14772. DOI:10.1039/c3ta13188b |
| [31] |
T. Wu, P. Wang, J. Qian, et al., Dalton Trans. 46 (2017) 13793-13801. DOI:10.1039/C7DT02929B |
| [32] |
J. Ran, T.Y. Ma, G. Gao, et al., Energy Environ. Sci. 8 (2015) 3708-3717. DOI:10.1039/C5EE02650D |
| [33] |
J. Zhang, M. Zhang, C. Yang, X. Wang, Adv. Mater. 26 (2014) 4121-4126. DOI:10.1002/adma.201400573 |
| [34] |
Y.P. Zhu, T.Z. Ren, Z.Y. Yuan, ACS Appl. Mater. Inter. 7 (2015) 16850-16856. DOI:10.1021/acsami.5b04947 |
| [35] |
T. Sano, S. Tsutsui, K. Koike, et al., J. Mater. Chem. A 1 (2013) 6489-6496. DOI:10.1039/c3ta10472a |
| [36] |
X. Du, G. Zou, Z. Wang, X. Wang, Nanoscale 7 (2015) 8701-8706. |
| [37] |
H. Yu, R. Shi, Y. Zhao, et al., Adv. Mater 29 (2017) 160548. |
| [38] |
V.W. Lau, V.W. Yu, F. Ehrat, et al., Adv. Energy Mater 7 (2017) 1602251. DOI:10.1002/aenm.201602251 |
| [39] |
J. Ran, T.Y. Ma, G. Gao, et al., Energy Environ. Sci. 8 (2015) 3708-3717. DOI:10.1039/C5EE02650D |
| [40] |
C. Zhou, R. Shi, T. Zhang, et al., Nano Res. 11 (2018) 3462-3468. DOI:10.1007/s12274-018-2003-2 |
| [41] |
Y. Cui, Z. Ding, X. Fu, X. Wang, Angew. Chem. Int. Ed. 51 (2012) 11814-11818. DOI:10.1002/anie.201206534 |
| [42] |
X. Guan, Y. Ma, H. Li, et al., J. Am. Chem. Soc. 140 (2018) 4494-4498. DOI:10.1021/jacs.8b01320 |
| [43] |
S.J. Makowski, P. Köstler, W. Schnick, Chem. Eur. J. 18 (2012) 3248-3257. DOI:10.1002/chem.201103527 |
| [44] |
F. Dong, L. Wu, Y. Sun, et al., J. Mater. Chem. 21 (2011) 15171-15174. DOI:10.1039/c1jm12844b |
| [45] |
Z. Zhang, K. Leinenweber, M. Bauer, et al., J. Am. Chem. Soc. 123 (2001) 7788-7796. DOI:10.1021/ja0103849 |
| [46] |
D.T. Vodak, K. Kim, L. Iordanidis, et al., Chem.-Eur. J. 9 (2003) 4197-4201. DOI:10.1002/chem.200304829 |
| [47] |
J.P. Zou, L.C. Wang, J. Luo, et al., Appl. Catal. B:Environ. 193 (2016) 103-109. |
| [48] |
J. Zhang, F. Guo, X. Wang, Adv. Funct. Mater. 23 (2013) 3008-3014. DOI:10.1002/adfm.201203287 |
| [49] |
S. Yan, Z. Li, Z. Zou, Langmuir 25 (2009) 10397-10401. DOI:10.1021/la900923z |
| [50] |
H. Zhao, X. Ding, C. Wang, et al., Sci. Bull. 62 (2017) 602-609. DOI:10.1016/j.scib.2017.03.005 |
| [51] |
A. Halder, S. Karak, M. Addicoat, et al., Angew. Chem. Int. Ed. 130 (2018) 5899-5904. DOI:10.1002/ange.201802220 |
| [52] |
H. Yu, R. Shi, T. Zhang, et al., Adv. Mater 29 (2017) 1605148. DOI:10.1002/adma.201605148 |
| [53] |
N. Boonprakob, N. Wetchakun, S. Phanichphant, et al., J. Colloid. Interf. Sci. 417 (2014) 402-409. DOI:10.1016/j.jcis.2013.11.072 |
| [54] |
X. Zhou, F. Peng, H. Wang, et al., Chem. Commun. 47 (2011) 10323-10325. DOI:10.1039/c1cc13862f |
| [55] |
V. Štengl, S. Bakardjieva, T.M. Grygar, et al., Chem. Cent. J. 7 (2013) 41-42. DOI:10.1186/1752-153X-7-41 |
| [56] |
H. Wang, X. Yuan, Y. Wu, et al., Appl. Catal. B:Environ. 174 (2015) 445-454. |
| [57] |
W. Yan, Y. Yu, K. Ding, et al., Solar RRL 2 (2018) 1800058. DOI:10.1002/solr.201800058 |
| [58] |
M.S. Lee, D.R. Whang, H.J. Choi, et al., Carbon 122 (2017) 515-523. DOI:10.1016/j.carbon.2017.07.001 |
| [59] |
J.P. Zou, S.L. Luo, L.Z. Zhang, et al., Appl. Catal. B:Environ. 140 (2013) 608-618. |
| [60] |
J. Ding, Z. Dai, F. Qin, et al., Appl. Catal. B:Environ. 205 (2017) 281-291. DOI:10.1016/j.apcatb.2016.12.018 |
| [61] |
G. Dong, K. Zhao, L. Zhang, Chem. Commun. 48 (2012) 6178-6180. DOI:10.1039/c2cc32181e |
| [62] |
F. Fina, H. Menard, J.T. Irvine, Phys. Chem. Chem. Phys. 17 (2015) 13929-13936. DOI:10.1039/C5CP00560D |
| [63] |
P. Bera, K. Priolkar, A. Gayen, et al., Chem. Mater. 15 (2003) 2049-2060. DOI:10.1021/cm0204775 |