 2020, Vol. 31
2020, Vol. 31 


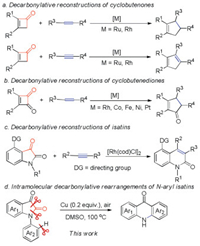
The reconstruction of new carbon skeletons after transition-metal-catalyzed decarbonylation of cycloketones provides rapid, atom- and step economical synthetic approaches to novel organic molecules that cannot be obtained by a simple combination of traditional synthetic methods (Scheme 1).

|
Download:
|
| Scheme 1. Decarbonylation and reconstructions of cycloketones. | |
{kind=link}
Highly strained four-membered ring ketones such as cyclo-butenones and cyclobutenediones have been recognized as versatile reagents for the construction of various multicyclic compounds through decarbonylative coupling with alkenes or alkynes (Schemes 1a and b) [1a-d]. The intermolecular decarbonylative couplings of cyclobutenones and cyclobutenediones were first reported by Kondo/Mitsudo [1a, c]. Compared to highly strained four-membered ring ketones, the decarbonylative reconstruction of a less-strained system, such as five- or six-membered ring ketones, has more challenges. Recently, Dong reported an intriguing decarbonylative C—C activation/2π-insertion reaction that applied the less strained five-membered ring diketones isatins as the substrates (Scheme 1c) [1e]. Cu(I) and Cu(II) salts-catalyzed ring-opening of N-aryl isatins generated in situ leading to acridones were reported by Zhou, Fu and Zhang [2, 3]. Herein, we first report an intramolecular Cu(0) powder-catalyzed decarbonylative rear-rangement reaction of N-aryl isatins leading to various acridones aided by DMSO under air atmosphere, in which a catalytic amount of sole and low-cost catalyst Cu(0) powder was used, without using basic and acidic conditions, additives and noble-catalysts (Scheme 1d).
Acridone motif is encountered in many natural products, dyes and pigments, bioactive molecules, fluorescent labels, and functional materials [4–7]. Acridones could be prepared from the corresponding N-aryl-substituted acids [4a, b], amides [8] and ketones [2] by C—C bond formation, or from diphenylamines via the palladium/copper co-catalyzed oxidative carbonylation [9]. Acridones have also been prepared from 2-aminobenzophenones via C—H amination [10]. As the substrates in the above intramolecular annulation methods require prior preparation, their applications are limited. Other routes proceeding through one-pot intermolecular annulation reactions to prepare acridones have been reported [3, 11], which have the advantages of step-economy and a high reaction efficiency. Although these methods provide a readily access to substituted acridones, they have some drawbacks, such as the use of inaccessible or expensive substrates, relatively harsh and strict reaction conditions, excessive additives. Therefore, the design of a greener and practical strategy for acridone preparation from readily available substrates would be highly interesting.
Indoline-2, 3-diones (isatins) are versatile synthetic blocks for synthesis of a vast array of biologically active compounds, naturally occurring alkaloids, medicinal agents and other functional molecules [12]. The readily availability and versatile reactivity of isatins [13], and the non-toxicity and low-cost of green DMSO [14] prompted us to develop a simple and greener synthetic method of acridones.
After extensive screening of different reaction parameters, optimum conditions were identified to be Cu(0) powder (0.2 equiv.) as a catalyst, and DMSO as a solvent at 100 ℃ under an air atmosphere (entry 3, Table S1 in Supporting information).
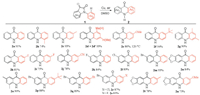
With these optimized conditions in hand, we proceeded to evaluate the scope of this decarbonylative rearrangement reaction, and the results summarized in Scheme 2.

|
Download:
|
| Scheme 2. Extension of reaction scope. Reaction conditions: 1a (0.2 mmol), Cu (0.2 equiv.), DMSO (2 mL), air, 100 ℃., 24–36 h. | |
{kind=link}
These transformations tolerated both electron-donating groups (EDG) and electron-withdrawing groups (EWG) on the benzene rings of both isatin moiety (Ar1) and N-aryl moiety (Ar2) to form various acridones 2 in good to excellent yields. N-Aryl isatins (1c–1g) bearing EDGs on the benzene ring of Ar2 showed higher reactivities and afforded a little bit higher yields than those bearing EWGs (1h–1j). The reaction of 1b bearing an ortho-methyl gave a lower yield compared with those bearing meta-and para-methyl groups (1b vs. 1c and 1d), owing to steric hindrance in the rearrangement reaction caused by ortho-substituent. Notably, the meta-Me-substituted substrate (1d) showed interesting regioselectivity, affording a product mixture with a 1:19 ratio in 93% yield (2d/2d'), which indicated that the recyclization at the para-position relative to methyl group was preferable to that at the ortho-position, based on the ratio of the isolated regioisomers. The electronic nature of substituents on the benzene ring of Ar1 had no evident influence on the reactions (1k–1m vs. 1n and 1o). Next, we examined the decarbonylative rearrangement reactions of N-aryl isatins (1p–1u) having various substituents including EDGs and EWGs on both benzene rings of Ar1 and Ar2. Noteworthily, these reaction substrates afforded the corresponding acridones (2p–2u) in high yields, as well.
To gain insight into the mechanism of this reaction, free radical trapping experiments were conducted (Eq. 1, Scheme S1 in Supporting information). It was found that compound 2a could be isolated in 90%, 89%, 91%, 83% yields in the presence of 2, 2, 6, 6-tetramethyl-1-piperidinyloxy (TEMPO, 1.0 equiv.), butylated hydroxytoluene (BHT, 1.0 equiv.) and N-tert-butyl-α-phenylnitrone (PBN, 1.0 equiv. and 5.0 equiv.), respectively, under the optimized conditions, indicating free radical process might not be involved in these transformations.
The catalyst Cu(0) powder was purchased from commercial sources and analyzed by powder X-ray diffraction (XRD, Fig. S1 in Supporting information) which showed the reagent is pure. Then, three blank experiments performed without 1a in the presence of 0.2 mmol of Cu(0) powder in DMSO (10 mL) at 100 ℃ for 24 h under N2, and air atmosphere conditions, respectively, to investigate the variation of Cu species. The black powders obtained by filtration at the end of these reactions, were analyzed by powder X-ray diffraction (Figs. S2 and S3 in Supporting information). XRD showed that the Cu species components of these black powders were 52.4% Cu(0) /47.6% Cu2O/0.0% CuO, and 5.7% Cu(0) /29.8% Cu2O/64.5% CuO, respectively. Cu2O and CuO were detected at air atmosphere, indicating that these decarbonylative rearrangement reactions might be catalyzed by in situ generated Cu2O, and CuO. Therefore, two reactions were conducted under the optimized conditions using Cu2O, and CuO instead of Cu (0) powder (Eqs. 2 and 3, Scheme S4 in Supporting information). In fact, Cu2O and CuO were effective for the transformations, however, gave lower yields in longer times. These results illustrated that Cu(0) might be a more rational catalytic species than Cu2O and CuO under current conditions. This was further confirmed by another control experiment (Eq. 4), which was conducted under N2 atmosphere using a higher temperature 130 C and greater amount of Cu(0) powder (0.5 equiv.) for 24 h to afford product 2a in 93% yield. XRD showed that the black powders obtained at the end of the reaction was Cu(0) powder (no Cu(I) and Cu(II) salts detected, Fig. S4 in Supporting information), indicating the catalytic Cu(0) species is conceivable.
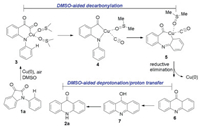
After many attempts, no intermediate generated in the rear-rangement processes could be characterized. To better understand the mechanism of this transformation, density functional theory (DFT) [15] calculations were carried out (Fig. 1). The possible mechanistic pathway of Cu(0) should consist of five important elementary processes: (1) oxidative addition, (2) decarbonylation, (3) metal migration, (4) redutive elimination, and (5) hydrogen transfer. In the reaction, the solvent DMSO would be ligand for Cu. Firstly, oxidative addition process has three kinds of possible adducts, where C1—C2 bond, C2—C3 bond, and C3—N bond in the heterocyclic ring can participate in coordination. The calculated activation energy data indicated that C3—N bond activation was the lowest (11.1 kcal/mol), and the afforded intermediate 3 was the most stable. The following decarbonylation process required 10.5 kcal/mol free energy to form intermediate 4. Then, the Cu migration happened and afforded seven-membered ring intermediate 5 by overcoming an 18.7 kcal/mol barrier. The further reductive elimination proceeded by climbing 20.5 kcal/mol activation energy to form intermediate 6. Finally, the product 2a is formed by two proton transfer processes. The activation energies of direct proton transfer were high (47.1 kcal/mol for TS5-1, 32.9 kcal/mol for TS7-1 and 39.2 kcal/mol for TS8-1).

|
Download:
|
| Fig. 1. Energy profile of the reaction catalyzed by Cu(0). | |
{kind=link}
Based on the results of the optimization experiment and reports about solvent-aided proton tranfer [16], therefore, we proposed that the solvent DMSO may play a crucial role in this process. Further computational calculations were performed using DMSO as a model solvent to elucidate the reason. As expected, DMSO is indeed crucial for the proton transfer. Specifically, the formation of 8 from 6 through by DMSO-aided TS5 only need 5.8 kcal/mol, and the transformation from intermediate 8 to 2a only by overcoming 21.7 kcal/mol with the assistance of DMSO. Moreover, activating the C—H bond of the N-phenyl ring by Cu(0) was also calculated, and the energy barrier of C–H activation step is too high (53.3 kcal/mol) (Fig. S5 in Supporting information). Similarly, after activating C—H bond by CuO, direct intramolecular nucleophilic attack at the ketone to form tricyclic intermediate 12 also required a high energy 52.0 kcal/mol (Fig. S5).
On the basis of the experimental results and DFT calculations, the mechanism of the decarbonylative rearrangement reaction for the formation of acridones 2 is described in Scheme 3. Under the applied reaction conditions, the amide C—N bond of 1a was cleaved through Cu insertion to produce a Cu complex 3 with the assist of DMSO. The metallacycle 3 underwent decarbonylation and Cu migration to form a seven-membered metallacycle 5. The intermediate 6 was then generated by a reductive elimination, which further converted to the more stable intermediate 8 with the assistant of DMSO followed by DMSO-aided deprotonation and proton transfer leading to the final product 2a.

|
Download:
|
| Scheme 3. Proposed mechanism. | |
{kind=link}
In summary, we have developed a Cu(0)-catalyzed decarbonylative rearrangement reaction through DMSO-aided catalytic carbonyl activation of isatins followed by intramolecular CO extrusion, Cu migration, reductive elimination and DMSO-aided proton migration processes. This reaction provides an attractive alternative in terms of green chemistry to construct various biologically interesting acridones from readily available N-aryl isatins in high to excellent yields, and show good functional group tolerance and wide substrate scope. DFT calculations reveal that multiple DMSO-assisted decarbonylation, deprotonation and proton transfer are crucial for the success of the transformation.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Natural Science Foundation of China (Nos. U1604285 and 21877206), the Program for Changjiang Scholars and Innovative Research Team in University of China (No. IRT1061), and the 111 Project (No. D17007).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.043.
| [1] |
(a) T. Kondo, Y. Taguchi, Y. Kaneko, M. Niimi, T. Mitsudo, Angew. Chem. Int. Ed. 43 (2004) 5369-5372; (b) P. Chen, T. Xu, G. Dong, Angew. Chem. Int. Ed. 53 (2014) 1674-1678; (c) T. Kondo, A. Nakamura, T. Okada, et al., J. Am. Chem. Soc. 122 (2000) 6319-6320; (d) M. Murakami, H. Amii, K. Shigeto, Y. Ito, J. Am. Chem. Soc. 118 (1996) 8285-8290; (e) R. Zeng, G. Dong, J. Am. Chem. Soc. 137 (2015) 1408-1411. |
| [2] |
(a) W. Zhou, Y. Yang, Y. Liu, G.J. Deng, Green Chem. 15 (2013) 76-80; (b) J. Yu, H. Yang, Y. Jiang, H. Fu, Chem. -Eur. J. 19 (2013) 4271-4277. |
| [3] |
H. Wu, Z. Zhang, Q. Liu, et al., Org. Lett. 20 (2018) 2897-2901. DOI:10.1021/acs.orglett.8b00957 |
| [4] |
(a) I.B. Taraporewala, J.W. Cessac, T.C. Chanh, A.V. Delgado, R.F. Schinazi, J. Med. Chem. 35 (1992) 2744-2752; (b) O. Tabarrini, G. Manfroni, A. Fravolini, et al., J. Med. Chem. 49 (2006) 2621-2617; (c) J.X. Kelly, M.J. Smilkstein, R. Brun, et al., Nature 459 (2009) 270-273. |
| [5] |
T. Faller, K. Hutton, G. Okafo, et al., Chem. Commun. (1997) 1529-1530. DOI:10.1039/A701787A |
| [6] |
D. Zhang, X. Jiang, H. Yang, et al., Org. Biomol. Chem. 11 (2013) 3375-3381. DOI:10.1039/c3ob27500k |
| [7] |
(a) X. Ye, P.N. Plessow, M.K. Brinks, et al., J. Am. Chem. Soc. 136 (2014) 5923-5929; (b) L.A. Graham, J. Suryadi, T.K. West, G.L. Kucera, U. Bierbach, J. Med. Chem. 55 (2012) 7817-7827. |
| [8] |
S.L. MacNeil, B.J. Wilson, V. Snieckus, Org. Lett. 8 (2006) 1133-1136. DOI:10.1021/ol053162e |
| [9] |
J. Wen, S. Tang, F. Zhang, R. Shi, A. Lei, Org. Lett. 19 (2017) 94-97. DOI:10.1021/acs.orglett.6b03356 |
| [10] |
(a) W. Zhou, Y. Liu, Y. Yang, G.J. Deng, Chem. Commun. 48 (2012) 10678-10680; (b) P.C. Huang, K. Parthasarathy, C.H. Cheng, Chem. -Eur. J. 19 (2013) 460-464. |
| [11] |
(a) J. Zhao, R.C. Larock, J. Org. Chem. 72 (2007) 583-588; (b) X. Pang, Z. Lou, M. Li, L. Wen, C. Chen, Eur. J. Org. Chem. 15 (2015) 3361-3369. |
| [12] |
(a) Y. Koguchi, J. Kohno, M. Nishio, et al., J. Antibiot. 53 (2000) 105; (b) M. Somei, F. Yamada, Nat. Prod. Rep. 20 (2003) 216-242; (c) C. Jing, T. Shi, D. Xing, X. Guo, W.H. Hu, Green Chem. 15 (2013) 620-624; (d) E.C. Elliott, E.R. Bowkett, J.L. Maggs, et al., Org. Lett. 20 (2011) 5592-5596; (e) P. Wu, H. Gao, J. Sun, C.G. Yan, Chin. Chem. Lett. 28 (2017) 329-322; (f) K. Stratmann, R.E. Moore, R. Bonjouklian, et al., J. Am. Chem. Soc. 116 (1994) 9935-9942; (g) J.L. Jimenez, U. Huber, R.E. Moore, G.M.L. Patterson, J. Nat. Prod. 62 (1999) 569-572; (h) H.B. Rasmussen, J.K. Macleod, J. Nat. Prod. 60 (1997) 1152-1154; (i) T. Tokunaga, W.E. Hume, T. Umezome, et al., J. Med. Chem. 44 (2001) 4641-4649; (j) Z. Xu, S. Zhang, C. Gao, et al., Chin. Chem. Lett. 28 (2017) 159-167; (k) R. Shintani, M. Inoue, T. Hayashi, Angew. Chem. Int. Ed. 45 (2006) 3353-3356; (l) R. Zeng, G. Dong, J. Am. Chem. Soc. 137 (2015) 1408-1411; (m) K. Meena, S. Kumari, J.M. Khurana, et al., Chin. Chem. Lett. 28 (2017) 136-142; (n) R.G. Shi, C.G. Yan, Chin. Chem. Lett. 27 (2016) 575-578. |
| [13] |
(a) T. Liu, H. Yang, Y. Jiang, H. Fu, Adv. Synth. Catal. 355 (2013) 1169-1176; (b) G.C. Senadi, W.P. Hu, S.S.K. Boominathan, J.J. Wang, Chem. Eur. J. 21 (2015) 998-1003; (c) P. Moser, A. Sallmann, I. Wiesenberg, J. Med. Chem. 33 (1990) 2358-2368; (d) S. Nizalapur, O. Kimyon, N.N. Biswas, et al., Org. Biomol. Chem. 14 (2016) 680-693; (e) P.C. Huang, P. Gandeepan, C.H. Cheng, Chem. Commun. 49 (2013) 8540-8542; (f) J. Du, Y. Yang, H. Feng, Y. Li, B. Zhou, Chem. -Eur. J. 20 (2014) 5727-5731; (g) P. Qian, J.H. Su, Y. Wang, et al., J. Org. Chem. 82 (2017) 6434-6440; (h) A. Ilangovan, G. Satish, Org. Lett. 15 (2013) 5726-5729; (i) C. Zhang, S. Li, F. Buress, et al., ACS Catal. 6 (2016) 6853-6860; (j) Y. Zi, Z.J. Cai, S.Y. Wang, S.J. Ji, Org. Lett. 16 (2014) 3094-3097; (k) J. Luo, S. Gao, Y. Ma, G. Ge, Synlett 29 (2018) 969-973; (l) B. Tang, R. Song, C. Wu, et al., J. Am. Chem. Soc. 132 (2010) 8900-8902. |
| [14] |
(a) T.X. Liu, S. Yue, C. Wei, et al., Chem. Commun. 54 (2018) 13331-13334; (b) S. Song, X. Li, X. Sun, Y. Yuan, N. Jiao, Green Chem. 17 (2015) 3285-3289; (c) Z. Zhang, Q. Tian, J. Qian, et al., J. Org. Chem. 79 (2014) 8182-8188; (d) J. Qian, Z. Zhang, Q. Liu, T. Liu, G. Zhang, Adv. Synth. Catal. 356 (2014) 3119-3124. |
| [15] |
M.J. Frisch, G.W. Trucks, H.B. Schlegel, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013.
|
| [16] |
(a) Q.C. Zhang, X. Li, X. Wang, et al., Org. Chem. Front. 6 (2019) 679-687; (b) X. Li, S.J. Li, Y. Wang, et al., Catal. Sci. Technol. 9 (2019) 2514-2522. |