 2020, Vol. 31
2020, Vol. 31 


b College of Life Science & Bioengineering, Beijing University of Technology, Beijing 100124, China;
c Department of Organic Chemistry, Faculty of Chemistry, Beijing University of Chemical Technology, Beijing 100029, China
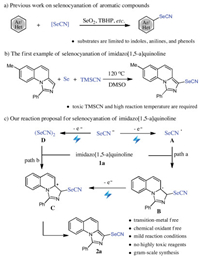
Imidazo[1, 5-a]quinolines are recognized as ubiquitous struc-tural motifs that existed widely in drug-relevant molecules and biologically active agents [1-3]. Due to the great potential of these compounds, many synthetic methodologies have been developed for the construction of imidazo[1, 5-a]quinolines [4-8]. Meanwhile, organic selenocyanates displayed a wide range of biological activities, such as anti-cancer and androgen receptor inhibition activities [9, 10]. Moreover, organic selenocyanates are also synthetically useful reagents which can be transformed into other selenium-containing organic compounds (selenols, diselenides, etc.) [11-13]. In this context, the introduction of selenocyanates into imidazo[1, 5-a]quinolines is of interest for synthetic and medicinal chemists. In contrast with numerous practical methods for selenation of aromatic compounds with diselenides (ArSeSeAr) [14-16], the selenocyanation of aromatic compounds continues to be underdeveloped in spite of undisputable advances have been achieved (Scheme 1a) [17, 18]. First, the developed synthetic methodologies are largely restricted to the use of stoichiometric amounts of toxic chemical oxidants such as SeO2 and TBHP. Second, the aromatic compounds are limited to reactive electron-rich systems such as indoles and aniline derivatives. However, the selenocyanation of other heterocyclic aromatic compounds is less explored. Recently, Feng, Yan and co-workers reported the first example of the selenocyanation of imidazo[1, 5-a]quinolines with high efficiency (Scheme 1b) [19]. However, excess amounts of toxic Se and TMSCN and the high reaction temperature are essential to the reaction process. Considering the synthetic and biological importance of imidazo[1, 5-a]quinoline scaffolds and organic selenocyanates, the development of a low-cost and eco-friendly C–H selenocyanation of imidazo[1, 5-a]quinolines in a regioselective manner is still desired.

|
Download:
|
| Scheme 1. Synthetic strategies for the selenocyanation of aromatic compounds. | |
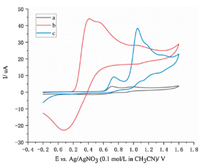
Organic electrochemistry has recently witnessed a renaissance by employing electrons as the "reagent" instead of toxic chemical oxidants or reductants, thus provides a sustainable and attractive way for the functionalization of organic molecules [20-26]. Inspired by the recent elegant examples of electrochemical selenation of unsaturated systems with ArSeSeAr independently developed by Sun [27], Kim [28], Guo [29] and Lei groups [30], we proposed an oxidative reaction sequence to realize the electrochemical C–H selenocyanation of imidazo[1, 5-a]quinolines (Scheme 1c). First, SeCN- is oxidized at the anode to give radical A. Subsequently, radical A has a radical addition to imidazo[1, 5-a]quinoline 1a giving radical B, which could be stabilized by the adjacent nitrogen atom. Then, radical B loses one electron to generate the carbocation C, which has a H+ elimination to afford the desired selenocyanated imidazo[1, 5-a]quinoline 2a. As a complementary reaction pathway, radical A may have a dimerization to give electrophilic selenocyanogen D, which has an electrophilic attack on imidazo[1, 5-a]quinoline affording the carbocation C. While the above-mentioned reaction pathways appear reasonable, cyclic voltammetric (CV) studies further confirmed the possibility of the proposed reaction pathways (Fig. 1). First, the CV of KSeCN in CH3CN exhibits two partially resolved oxidation processes at potentials between 0.4 V and 0.6 V, and a reduction process at 0.1 V, which correspond to the pseudohalide system of {SeCN--(SeCN)3--(SeCN)2} (curve b) [31, 32]. The observed irreversible process may result from the formation of (SeCN)2. Second, SeCN- is much easier to be oxidized to give reactive species than 1a (curve c), which is beneficial to regioselective C–H selenocyanation. In continuation of our interest in imidazo[1, 5-a]quinolines syntheses [4, 5, 7] and electrochemical C–heteroatom bonds formations [33-37], we herein report the first example of electrochemical C–H selenocyanation of imidazo[1, 5-a]quinolines. Under catalyst- and chemical oxidant-free conditions, synthetically and biologically important selenocyanated imidazo [1, 5-a]quinolines were obtained in good to excellent yields with cheap graphite and Ni plates as the electrodes. The gram-scale synthesis was also successfully conducted, which might demonstrate the potential value of this electrochemical selenocyanation protocol.

|
Download:
|
| Fig. 1. CV of related compounds in 0.1 mol/L n-Bu4NBF4/CH3CN using Pt wire and Ag wire as the working and counter electrodes, and Ag/AgNO3 (0.1 mol/L in CH3CN) as the reference electrode, scan rate: 100 mV/s, (a) blank (b) KSeCN (5 mmol/L), (c) 1a (5 mmol/L). | |
Initially, we took the selenocyanation of 1a with KSeCN as the model reaction to optimize the reaction conditions (Table 1). When the reaction was carried out in an undivided cell with Ni and graphite plates as the electrodes and n-Bu4NBF4/CH3CN as the electrolyte, the corresponding selenocyanated product 2a was obtained in 86% yield (entry 1). Other supporting electrolytes such as LiClO4 and n-Bu4NPF6 gave lower yields (entries 2 and 3). Further optimization showed that the electrode materials have much influence on the yield. For example, when C/Pt or Pt/Pt were used as the electrodes, the yield of 2a decreased to 41% and 47%, respectively (entries 4 and 5). However, when C/C were employed as the electrodes, only trace amount of product was observed (entry 6). The solvent optimizations showed that CH3CN was the optimal solvent, while other solvents such as MeOH, DMF, and DMSO gave much lower yields (entries 7–9). The control experiments showed that the electricity and TFA were essential for the transformation (entries 10 and 11). TFA not only helps the cathodic H2 revolution but also facilitates the Minisci-type radical substitution [38, 39]. Replacing TFA with HOAc decreased the yield of 2a to 31% (entry 12).
|
|
Table 1 Optimization of reaction condition.a |

{kind=link}
{kind=link}
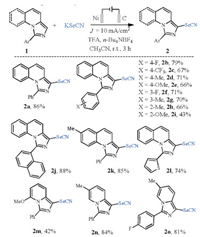
With the optimized conditions identified, the substrate scope of aryl-substituted imidazo[1, 5-a]quinolines was examined. As shown in Scheme 2, a series of selenocyanated imidazo[1, 5-a]quinolines were obtained in good to excellent yields. The electronic nature of the aryl substituents has an important role on the yield. For example, the presence of a strong electron-donating substituent lowered the yields (2d, 2e, 2g-2i), while the weak electron-withdrawing substituents could maintain the yields (2b, 2c, and 2f). The strong electron-donating substituents lowered the oxidation potentials of imidazo[1, 5-a]quinolines which would lead to the competition reaction with the SeCN- oxidation. This finding suggests that the electrophilic addition of selenocyanogen D to imidazo[1, 5-a]quinoline 2a affording intermediate C was the minor reaction pathway (Scheme 1c, path b). The naphthyl-substituted substrate was well tolerated under the optimized conditions, giving the corresponding product 2j in 88% yield. It is worth mentioning that thienyl-substituted substrate was smoothly selenocyanated affording the corresponding product 2l in 74% yield. For the imidazo[1, 5-a]pyridines, the selenocyanation reactions also underwent smoothly to give 2m-2o with up to 84% yield.

|
Download:
|
| Scheme 2. The substrate scope of aryl-substituted imidazo[1, 5-a]quinolones. Reaction conditions: 1 (0.3 mmol), KSeCN (0.75 mmol), TFA (0.7 mmol), 0.2 mol/L n-Bu4NBF4 in CH3CN (10 mL), Ni plate (1×2 cm2), graphite plate (1×2 cm2), J =10 mA/cm2, r.t., 3 h; isolated yield. | |
{kind=link}
Having established the substrate scope of aryl-substituted imidazo[1, 5-a]quinolines, the substrate scope of alkyl- and allylic-substituted imidazo[1, 5-a]quinolines was then investigated under the optimized conditions with a higher electronic density (Scheme 3). First, imidazo[1, 5-a]quinolines with linear aliphatic chains were examined. The aliphatic chains with no functional groups were well tolerated, giving the corresponding products 3a-3d with up to 85% yield. The benzyl-substituted imidazo[1, 5-a]quinoline was also a suitable substrate, albeit with a lower yield (3e). It is noteworthy that aliphatic chains with functionalities such as thioether and amides were also well tolerated, yielding functionalized imidazo[1, 5-a]quinolines 3f-3h in 54%–80% yields. The allylic substituted imidazo[1, 5-a]quinoline also underwent the selenocyanation reaction smoothly to give the corresponding product 3i in 35% yield. Next, the branched aliphatic substituents were also tested under the optimized conditions, and the corresponding selenocyanated imidazo[1, 5-a]quinolines 3j-3m were obtained with up to 83% yield. For the alkyl-substituted imidazo[1, 5-a]pyridine, the selenocyanation reaction also underwent smoothly to give the corresponding product 3n in 77% yield.

|
Download:
|
| Scheme 3. The substrate scope of alkyl-substituted imidazo[1, 5-a]quinolones. Reaction conditions: 1 (0.3 mmol), KSeCN (0.75 mmol), TFA (0.7 mmol), 0.2 mol/L n-Bu4NBF4 in CHCN (10 mL), Ni plate (1×2 cm2), graphite plate (1×2 cm2), J =12 mA/cm2, r.t., 3 h; isolated yield. | |
{kind=link}
To illustrate the synthetic utility of this protocol for the synthesis of selenocyanated imidazo[1, 5-a]quinolines, the reaction of 1g with KSeCN on a gram-scale was conducted (Scheme 4, details see the Supporting information). To our delight, the corresponding product 3g was obtained in 64% yield.

|
Download:
|
| Scheme 4. Gram-scale synthesis. | |
{kind=link}
To gain insight into the mechanism, the radical trapping experiment was carried out (Scheme 5). When butylated hydroxytoluene (BHT) as the radical scavenger was subjected into the reaction mixture, the yield of product 2a decreased to 56%. This result suggests that radical species were involved in this electrochemical selenocyanation transformation (Scheme 1c, path a). However, reaction pathway involving anion species could not be ruled out (Scheme 1c, path b). These results are in accord with the observed irreversible process of the pseudohalide system of {SeCN- -(SeCN)3- -(SeCN)2} (Fig. 1).

|
Download:
|
| Scheme 5. Radical-trapping experiment. | |
{kind=link}
In conclusion, we have developed the first example of electrochemical selenocyanation of imidazo[1, 5-a]quinolines with KSeCN under metal catalyst- and chemical oxidant-free conditions. This sustainable strategy shows a broad scope and great compatibility with functional groups, and affords synthetically and biologically important selenocyanated imidazo[1, 5-a]quinolines in good to excellent yields with cheap graphite and Ni plates as the electrodes. The gram-scale synthesis demonstrates the potential value of this electrochemical selenocyanation protocol.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful to the National Natural Science Foundation of China (Nos. 21602119, 21702013), the Foundation of He'nan Educational Committee (No. 16A150057), Program for Science and Technology Innovation Talents in Universities of Henan Province (No. 19HASTIT033), the Beijing Natural Science Foundation (No. 2184115) and the Fundamental Research Funds for the Central Universities (Nos. XK1802-6, buctrc201721).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.11.037.
| [1] |
C. Alcouffe, A. Badore, F. Bono, et al., US Patent, US 2008108648 A1, 2008.
|
| [2] |
M. Follmann, J.P. Stasch, G. Redlich, et al., US Patent, US 2014171434 A1, 2014.
|
| [3] |
A. Shvartsbart, S. Shepard, A.P. Combs, et al., US Patent, US 2017129899 A1, 2017.
|
| [4] |
H.Q. Wang, W.T. Xu, Z.Q. Wang, L.T. Yu, K. Xu, J. Org. Chem. 80 (2015) 2431-2435. DOI:10.1021/jo5027723 |
| [5] |
H.Q. Wang, W.T. Xu, L.L. Xin, et al., J. Org. Chem. 81 (2016) 3681-3687. DOI:10.1021/acs.joc.6b00343 |
| [6] |
Z.D. Tan, H. Zhao, C.J. Zhou, H.F. Jiang, M. Zhang, J. Org. Chem. 81 (2016) 9939-9946. DOI:10.1021/acs.joc.6b02117 |
| [7] |
C.T. Feng, H.J. Wei, J. Li, P. Ya, K. Xu, Adv. Synth. Catal. 360 (2018) 4726-4730. DOI:10.1002/adsc.201801060 |
| [8] |
P. Qian, Z.H. Zhou, K.F. Hu, et al., Org. Lett. 21 (2019) 6403-6407. DOI:10.1021/acs.orglett.9b02317 |
| [9] |
N.D. Facompre, K. El-Bayoumy, Y.W. Sun, J.T. Pinto, R. Sinha, Cancer Prev. Res. 3 (2010) 975-984. DOI:10.1158/1940-6207.CAPR-10-0054 |
| [10] |
S.S. Mati, S.S. Roy, S. Chall, S. Bhattacharya, S.C. Bhattacharya, J. Phys. Chem. B 117 (2013) 14655-14665. DOI:10.1021/jp4090553 |
| [11] |
A. Krief, W. Dumont, C. Delmotte, Angew. Chem. Int. Ed. 39 (2000) 1669-1672. DOI:10.1002/(SICI)1521-3773(20000502)39:9<1669::AID-ANIE1669>3.0.CO;2-6 |
| [12] |
J.C. Guillemin, G. Bajor, E.H. Riague, B. Khater, T. Veszprémi, Organometallics 26 (2007) 2507-2518. DOI:10.1021/om061067j |
| [13] |
Y. Guan, S.D. Townsend, Org. Lett. 19 (2017) 5252-5255. DOI:10.1021/acs.orglett.7b02526 |
| [14] |
K. Sun, X. Wang, Y.H. Lv, et al., Chem. Commun. 52 (2016) 8471-8474. DOI:10.1039/C6CC04225B |
| [15] |
C. Wu, H.J. Xiao, S.W. Wang, et al., ACS Sustainable Chem. Eng. 7 (2019) 2169-2175. DOI:10.1021/acssuschemeng.8b04877 |
| [16] |
V. Rathore, S. Kumar, Green Chem. 21 (2019) 2670-2676. DOI:10.1039/C9GC00007K |
| [17] |
A.V. Kachanov, O.Y. Slabko, O.V. Baranova, E.V. Shilova, V.A. Kaminskii, Tetrahedron Lett. 45 (2004) 4461-4463. DOI:10.1016/j.tetlet.2004.04.071 |
| [18] |
N. Muniraj, J. Dhineshkumar, K.R. Prabhu, ChemSelect 5 (2016) 1033-1038. DOI:10.1002/slct.201600292 |
| [19] |
C.T. Feng, Y. Peng, G.R. Ding, et al., Chem. Commun. 54 (2018) 13367-13370. DOI:10.1039/C8CC07905F |
| [20] |
S. Tang, Y.C. Liu, A.W. Lei, Chem 4 (2018) 27-45. DOI:10.1016/j.chempr.2017.10.001 |
| [21] |
C. Ma, P. Fang, T.S. Mei, ACS Catal. 8 (2018) 7179-7189. DOI:10.1021/acscatal.8b01697 |
| [22] |
N. Sauermann, T.H. Meyer, Y.A. Qiu, L. Ackermann, ACS Catal. 8 (2018) 7086-7103. DOI:10.1021/acscatal.8b01682 |
| [23] |
Y.Y. Jiang, K. Xu, C.C. Zeng, Chem. Rev. 118 (2018) 4485-4540. DOI:10.1021/acs.chemrev.7b00271 |
| [24] |
G.S. Sauer, S. Lin, ACS Catal. 8 (2018) 5175-5187. DOI:10.1021/acscatal.8b01069 |
| [25] |
Z.H. Ye, F.Z. Zhang, Chin. J. Chem. 37 (2019) 513-528. DOI:10.1002/cjoc.201900049 |
| [26] |
H.B. Zhao, P. Xu, J. Song, H.C. Xu, Angew. Chem. Int. Ed. 57 (2018) 15153-15156. DOI:10.1002/anie.201809679 |
| [27] |
X. Zhang, C.G. Wang, H. Jiang, L.H. Sun, Chem. Commun. 54 (2018) 8781-8784. DOI:10.1039/C8CC04543G |
| [28] |
Y.J. Kim, D.Y. Kim, Org. Lett. 21 (2019) 1021-1025. DOI:10.1021/acs.orglett.8b04041 |
| [29] |
J.W. Hua, Z. Fang, J. Xu, et al., Green Chem. 21 (2019) 4706-4711. DOI:10.1039/C9GC02131K |
| [30] |
L. Sun, Y. Yuan, M. Yao, et al., Org. Lett. 21 (2019) 1297-1300. DOI:10.1021/acs.orglett.8b03274 |
| [31] |
M.E. Martins, C.E. Castellano, A.J. Calandra, A.J. Arvía, Anal. Chem. 50 (1978) 229-231. DOI:10.1021/ac50024a016 |
| [32] |
A. Solangi, A.M. Bond, L. Burgar, et al., J. Phys. Chem. B 115 (2011) 6843-6852. |
| [33] |
S. Zhang, F. Lian, M.Y. Xue, et al., Org. Lett. 19 (2017) 6622-6625. DOI:10.1021/acs.orglett.7b03333 |
| [34] |
S. Zhang, L.J. Li, M.Y. Xue, et al., Org. Lett. 20 (2018) 3443-3446. DOI:10.1021/acs.orglett.8b00981 |
| [35] |
S. Zhang, L.J. Li, H.Q. Wang, et al., Org. Lett. 20 (2018) 252-255. DOI:10.1021/acs.orglett.7b03617 |
| [36] |
H.Q. Wang, J.J. Zhang, J.J. Tan, et al., Org. Lett. 20 (2018) 2505-2508. DOI:10.1021/acs.orglett.8b00165 |
| [37] |
S. Zhang, L.J. Li, J.J. Zhang, et al., Chem. Sci. 10 (2019) 3181-3185. DOI:10.1039/C9SC00100J |
| [38] |
H. Ding, K. Xu, C.C. Zeng, J. Catal. 381 (2020) 38-43. DOI:10.1016/j.jcat.2019.10.030 |
| [39] |
H. Yan, Z.W. Hou, H.C. Xu, Angew. Chem. Int. Ed. 58 (2019) 4592-4595. DOI:10.1002/anie.201814488 |