 2020, Vol. 31
2020, Vol. 31 


b Guangdong Key Laboratory of Chiral Molecule and Drug Discovery, School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China;
c Guangdong Institute of Analysis(China National Analytical Center, Guangzhou), Guangzhou 510070, China
Hydroborations of alkynes provide facile routes to alkenyl boron compounds [1]. Typically, the cis-adducts are preferentially formed due to the concerted nature of direct hydroboration with trivalent boranes and the cis-selective property of migratory insertion when transition metal is involved. Examples on trans-hydroboration, however, are rare [2], and largely rely on the use of specifically designed catalyst or the use of directing group.
Ynamides, as a special class of alkynes with a nitrogen atom bonded directly to a sp-hybridized carbon, show intriguing reactivity in numerous synthetic transformations [3]. The biased distribution of the p electrons also renders the high regioselectivity possible. Recently, we have been engaged in the hydrofuctionalization reactions of ynamide toward the synthesis of structurally complex molecules [4]. The hydroboration of ynamides, would deliver alkenes substituted with two valuable functional groups. The first example of hydroboration of ynamide was disclosed by Witulski in 2000 wherein N-sulfonyl ynamide reacts directly with catecholborane to give a β-E-vinylborane with exclusive region- and stereoselectivity (Scheme 1a) [5]. Only terminal ynamide was applicable and the product was found to be instable and difficult for isolation. In the aid of a zirconocene [6] or copper(I) [7] catalyst, the β-selective cis-hydroborations of internal ynamides was also recently realized (Schemes 1b and c). Interestingly, a similar copper(I) catalytic system comprising a different phosphine ligand reverses the regioselectivity to deliver an α-selective cis-adduct, as reported by Zhu (Scheme 1d) [8]. In spite of these elegant progresses, to date, no general methods on the β-selective trans-hydroboration of ynamides have been known from the literature, although the products from this process are useful synthons in organic synthesis.

|
Download:
|
| Scheme 1. Reported hydroboration of ynamides and inspiration of this work. | |
Different from the typical concerted hydroboration and organometallic hydroboration mechanism, we envisioned that a boryl radical addition pathway might be suited for a β-selective trans-hydroboration of ynamides. Recently, Lewis-base ligated boranes are demonstrated to be efficient boron sources for organoboron preparation [9]. Particularly important is the N-heterocyclic carbene (NHC)-boryl radicals, prepared via hydrogen abstraction from NHC-BH3 with different initiators, are known to undergo radical addition reactions to activated alkenes and alkynes [10]. Other inspiration comes from the elegant studies by Perez-Luna, who demonstrated the trans-selective radical silylzincation ynamides was efficient by using a combination of TMS3SiH/Et2Zn [11]. We thus reasoned a similar radical borylzincation pathway should be possible given the similarity of boryl-centered radical with sily-centered radicals in terms of bond dissociation energy (X-H) and polarity [12].
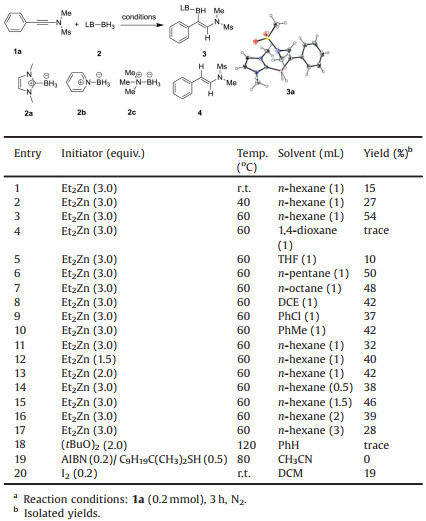
We commenced our studies by investigating the reaction of phenyl-substituted N-sulfonyl ynamide 1a with 1, 3-dimethylimidazol-2-ylidene borane 2a (2.0 equiv.) in the presence of 3.0 equiv. of Et2Zn in n-hexane [11a]. When the reaction was conducted at room temperature, incomplete conversion of the ynamide 1a was found (entry 1, Table 1). Nevertheless, the desired hydroboration product 3a was isolated in 15% yield as a single region- and stereoisomer. The structure of 3a was unambiguously confirmed by X-ray crystallographic analysis (CCDC Repository No. 1949342, See the Supporting information for details). Elevating the temperature to 60 ℃ led to an improved yield of 54% (entries 2 and 3). The trans-hydrogenation forming compound 4 (36% yield) was identified as the major side reaction. No improvement was accomplished by screening different solvents (entries 4–10). Further, the employment of different Et2Zn loadings (entries 11–13), or different reaction concentrations (entries 14–17) all failed to give a better yield. Surprisingly, the use of Curran's boryl radical addition conditions, featuring the use of di-tert-butyl peroxide as initiator, resulted in only trace amount of product (entry 18) [10c]. Also, the use of Wang's radical conditions (AIBN as initiator, RSH as hydrogen shuttle, CH3CN, 80 ℃) gave no desired product (entry 19) [10g]. Interestingly, the use of I2 as catalyst, which may form a borenium ion by reacting with NHC-BH3, also give the desired product, although in a low 19% yield (entry 20) [13]. Other Lewis-base ligated borane sources, such as pyridine-BH2 (2b) and Me3N-BH3 (2c) were also test, but no reactivity was found. Thus, the optimized reaction conditions were identified as: 3.0 equiv. of Et2Zn, 0.2 mol/L in n-hexane at 60 C for 3 h (entry 3).
|
|
Table 1 Reaction optimization. a |
{kind=link}
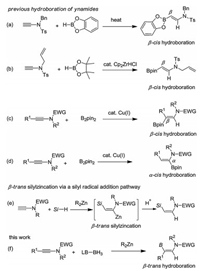
The scope and generality of this β-selective trans-hydroboration was then investigated. As shown in Scheme 2, aryl ynamides substituted with methyl (3b), ethyl (3c), tert-butyl (3d), fluoro (3e-g), chloro (3h), bromo (3i), CF3 (3j), and ester (3k, 3l) on the aryl ring all led to satisfactory yields. But the nitro functional group was not compatible well (3n), as significant amount of hydrogenation side product was formed. A thiophen-substituted substrate was also applicable for hydroboration (3o). Substrates with ortho-substitutent are potentially steric hindered, but their reactivity was not compromised (3g, 3h). In addition to aryl ynamides, alkyl ynamides underwent hydroboration as well (3p-r). It was found the reaction is quite sensitive to the protecting group on the nitrogen, as the displacement of Ms with Ts (1s, 1u, 1v), or Ns (1t), or the use of oxazolidinone derived (1w) ynamdies all shut down the reactivity completely.

|
Download:
|
| Scheme 2. Substrate scope. | |
{kind=link}
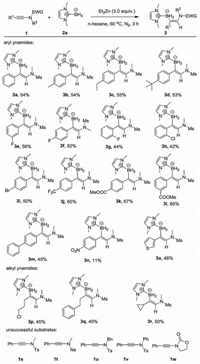
The mechanism of the reaction is worthy of discussion. According to the work of Perez-Luna, a mechanism involving the formation of boryl radical could be envisioned (Scheme 3a) [11a]. Firstly, a boryl radical is expected to be formed via the H atom transfer between NHC-BH3 and ethyl radical. This radical A then adds across the C≡C triple bond of ynamides. The regioselectivity of this step is in accordance to the addition reaction of silyl-centered radicals [11a]. The newly formed α-nitrogen-substituted vinylic radical B, could either react with Et2Zn by SH2 (hemolytic substitution) to form a vinylzinc intermediate C, or could directly abstract a hydrogen from the NHC-BH3 to deliver the final product. The protonation of the C-Zn bond in C during aqueous workup could also provide the hydroboration product.

|
Download:
|
| Scheme 3. Proposed reaction pathways. | |
{kind=link}
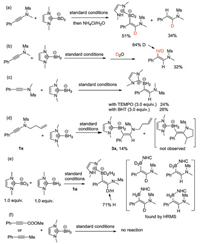
To distinguish these two pathways and explore the possibility of alternative reaction mechanism, a series of experiments were then conducted. First, the use of deuterated NHC-BD3 under the same reaction conditions led to a product with complete deuterium incorporation (Scheme 4a), which suggest the product is not derived from the protonation of vinylzinc C. The analysis of the side hydrogenation product concluded that the hydrogen α to the nitrogen comes from the NHC-BH3 as well (Scheme 4a), while the one β to the nitrogen is derived from water (Scheme 3b). The use of TEMPO or BHT as radical scavenger led to lower yields (Scheme 4b), but not as significant as observed by Curran in his NHC-boryl radical addition reaction [10d]. In addition, a but-3-en-1-yl substituted ynamide 1x gave a low yield of hydroboration due to low conversion (Scheme 4d). This may suggest the reaction is sensitive to steric hindrance. Further, the absence of cyclization product, together with results of the radical inhibition experiment, may rule out the formation of α vinylic radical (B, Scheme 3) in this reaction. Competition experiments with equal amount of NHC-BH3 and NHC-BD3 revealed a primary isotope effect value of 2.4 (Scheme 4e). HRMS analysis of the product showed the existence of four H/D scrambling compounds, thus excluding a concerted reaction mechanism. Prop-1-yn-1-ylbenzene and methyl 3-phe-nylpropiolate were two substrates applicable in Curran's radical trans-hydroboration of alkynes [10c]. However, no reactivity was observed when these two substrates were used in our protocol, suggesting the same boryl radical may not be formed (Scheme 4f).

|
Download:
|
| Scheme 4. Mechanistic studies. | |
{kind=link}
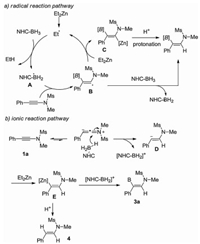
Based on these observations, although we cannot fully exclude the radical pathway at this time, an alternative polar mechanism is proposed (Scheme 3b). The polarization of the ynamide triple bond due to the electron-donation of nitrogen affords a keteniminium, which makes the α-carbon of ynamide electrophilic. Thus, the hydride transform from the NHC-BH3 is expected to occur regioselectively. This process would release a borenium ion, similar species has been previously proposed [14]. The vinyl anion D may be trapped rapidly with Et2Zn to form a β-vinylzinc adduct E, which then reacts with the previously formed borenium ion to deliver the hydroboration product. On the other hand, since the borenium ion is unstable and may be partly consumed via unproductive pathways, some amount of the vinylzinc E may remain untouched during the reaction. The protonation of E upon aquous workup then forms the side hydrogenation product. This mechanistic profile seems in better agreement with the mechanistic studies.
Finally, the synthetic transformation of the C B bond in the product was achieved via a palladium-catalyzed Suzuki-Miyaura coupling with aryl iodides, providing a simple and stereospecific route to multi-substituted enamides (Eq. 1).
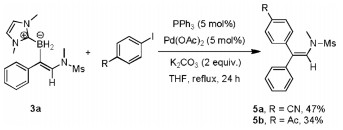
|
(1) |
In summary, we have developed an Et2Zn-promoted β- and trans-selective hydroboration of ynamides. The reaction is compatible with a series of aryl and alkyl substituents and provides the borylated enamides in generally acceptable yields. The derivatization of the product was demonstrated. The mechanism was discussed and a step-wise polar hydroboration pathway was suggested.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial supports from the Key Project of Chinese National Programs for Fundamental Research and Development (No. 2016YFA0602900), the Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (No. 2017BT01Y093), the Natural Science Foundation of China (No. 81873071), and the Pearl River S & T Nova Program of Guangzhou (No. 201906010059) are gratefully acknowledged.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.11.008.
| [1] |
(a) H.C. Brown, Organic Syntheses Via Boranes, Wiley, New York, 1975; (b) A. Pelter, K. Smith, H.C. Brown, Borane Reagents, Academic Press, New York, 1988; (c) M. Vaulter, G. Alcaraz, Science of Synthesis, Vinylboranes, Georg Thieme, (2014); (d) J. Chen, J. Guo, Lu Z, Chin. J. Chem. 36 (2018) 1075-1109. |
| [2] |
(a) T. Ohmura, Y. Yamamoto, N. Miyaura, J. Am. Chem. Soc. 122 (2000) 4990-4991; (b) C. Gunanathan, M. Hçlscher, F. Pan, W. Leitner, J. Am. Chem. Soc. 134 (2012) 14349-14352; (c) B. Sundararaju, A. Fgrstner, Angew. Chem. Int. Ed. 52 (2013) 14050-14054; (d) B. Sundararaju, A. Fgrstner, Angew. Chem. 125 (2013) 14300-14304; (e) Q. Wang, S.E. Motika, N.G. Akhmedov, J.L. Petersen, X. Shi, Angew. Chem. Int. Ed 53 (2014) 5418â€"5422; (f) Q. Wang, S.E. Motika, N.G. Akhmedov, J.L. Petersen, X. Shi, Angew. Chem.126 (2014) 5522-5526; (g) J.V. Obligacion, J.M. Neely, A.N. Yazdani, I. Pappas, P.J. Chirik, J. Am. Chem. Soc. 137 (2015) 5855-5858; (h) W.J. Jang, W.L. Lee, J.H. Moon, J.Y. Lee, J. Yun, Org. Lett.18 (2016) 1390-1393; (i) S. Xu, Y. Zhang, B. Li, S.Y. Liu, J. Am. Chem. Soc. 138 (2016) 14566-14569; (j) N. Gorgas, L.G. Alves, B. Stçger, et al., J. Am. Chem. Soc. 139 (2017) 8130-8133; (k) K. Yuan, N. Suzuki, S.K. Mellerup, et al., Org. Lett. 18 (2016) 720-723; (l) K. Nagao, A. Yamazaki, H. Ohmiya, M. Sawamura, Org. Lett. 20 (2018) 1861-1865; (m) J. Guo, B. Cheng, X. Shen, Z. Lu, J. Am. Chem. Soc. 139 (2017) 15316-15319. |
| [3] |
(a) K.A. DeKorver, H. Li, A.G. Lohse, et al., Chem. Rev. 110 (2010) 5064-5106; (b) G. Evano, A. Coste, K. Jouvin, Angew. Chem. Int. Ed. 49 (2010) 2840-2859; (c) P. Lu, Y. Wang, Chem. Soc. Rev. 41 (2012) 5687-5705; (d) X.N. Wang, H.S. Yeom, L.C. Fang, et al., Acc. Chem. Res. 47 (2014) 560-578; (e) A.M. Cook, C. Wolf, Tetrahedron Lett. 56 (2015) 2377-2392; (f) F. Pan, C. Shu, L.W. Ye, Org. Biomol. Chem. 14 (2016) 9456-9465; (g) X. Li, Y. Sun, L. Zhang, B. Peng, Chin. J. Org. Chem. 36 (2016) 2530-2544; (h) G. Duret, V. Le Fouler, P. Bisseret, V. Bizet, N.E. Blanchard, Eur. J. Org. Chem. 46 (2017) 6816-6830; (i) G. Evano, M. Lecomte, P. Thilmany, C. Theunissen, Synthesis 49 (2017) 3183-3214. |
| [4] |
(a) X.L. Han, C.J. Zhou, X.G. Liu, et al., Org. Lett. 19 (2017) 6108-6111; (b) J. Li, E. Lin, X.L. Han, Q. Li, H. Wang, Org. Lett. 21 (2019) 4255-4258; (c) P.P. Lin, X.L. Han, G.H. Ye, et al., J. Org. Chem. 84 (2019) 12966-12974. |
| [5] |
B. Witulski, N. Buschmann, U. Bergsträßer, Tetrahedron 56 (2000) 8473-8480. DOI:10.1016/S0040-4020(00)00773-0 |
| [6] |
R.W. Hoffmann, D. Brücknera, New J. Chem. 25 (2001) 369-373. DOI:10.1039/b009259m |
| [7] |
Y. Bai, F. Zhang, J. Shen, F. Luo, G. Zhu, Asian J. Org. Chem. 4 (2015) 626-629. DOI:10.1002/ajoc.201500119 |
| [8] |
G. He, S. Chen, Q. Wang, et al., Org. Biomol. Chem. 12 (2014) 5945-5953. DOI:10.1039/C4OB00979G |
| [9] |
J.M. Yang, Z.Q. Li, S.F. Zhu, Chin. J. Org. Chem. 37 (2017) 2481-2497. |
| [10] |
(a) D.P. Curran, A. Solovyev, M.M. Brahmi, Angew. Chem. Int. Ed. 50 (2011) 10294-10317; (b) T. Taniguchi, Eur. J. Org. Chem. 37 (2019) 6308-6319; (c) M. Shimoi, T. Watanabe, K. Maeda, D.P. Curran, T. Taniguchi, Angew. Chem., Int. Ed. 57 (2018) 9485-9490; (d) W. Dai, S.J. Geib, D.P. Curran, J. Am. Chem. Soc. 141 (2019) 12355-12361; (e) T. Watanabe, D. Hirose, D.P. Curran, T. Taniguchi, Chem. -Eur. J. 23 (2017) 5404-5409; (f) J. Qi, F.L. Zhang, Y.S. Huang, et al., Org. Lett. 20 (2018) 2360-2364; (g) S.C. Ren, F.L. Zhang, J. Qi, et al., J. Am. Chem. Soc. 139 (2017) 6050-6053; (h) J.K. Jin, F.L. Zhang, Q. Zhao, J.A. Lu, Y.F. Wang, Org. Lett. 20 (2018) 7558-7562; (i) M. Shimoi, K. Maeda, S.J. Geib, D.P. Curran, T. Taniguchi, Angew. Chem. Int. Ed. 58 (2019) 6357-6361; (j) S.C. Ren, F.L. Zhang, A.Q. Xu, et al., Nat. Commun. 10 (2019) 1934; (k) J.K. Jin, W.X. Zheng, H.M. Xia, F.L. Zhang, Y.F. Wang, Org. Lett. 21 (2019) 8414-8418; (l) X. Liu, E. Lin, G. Chen, et al., Org. Lett. 21 (2019) 8454-8458. |
| [11] |
(a) E. Romain, C. Fopp, F. Chemla, et al., Angew. Chem. Int. Ed. 53 (2014) 11333-11337; (b) K. Vega-Hernandez, E. Romain, A. Coffinet, et al., J. Am. Chem. Soc. 140 (2018) 17632-17642. |
| [12] |
(a) X. Pan, E. LacoÌ, te, J. Lalevee, D.P. Curran, J. Am. Chem. Soc.134 (2012) 5669-5674; (b) C. Chatgilialoglu, Chem. -Eur. J. 14 (2008) 2310-2320. |
| [13] |
(a) X. Pan, A. Boussonnière, D.P. Curran, J. Am. Chem. Soc. 135 (2013) 14433-14437; (b) J.E. Radcliffe, V. Fasano, R.W. Adams, P. Youa, M.J. Ingleson, Chem. Sci. 10 (2019) 1434-1441. |
| [14] |
(a) T.S. De Vries, A. Prokofjevs, E. Vedejs, Chem. Rev. 112 (2012) 4246-4282; (b) A. Solovyev, S.J. Geib, E. LacoÌ, te, D.P. Curran, Organometallics 31 (2012) 54- |