 2020, Vol. 31
2020, Vol. 31 


b Institute of Traditional Chinese Medicine & Natural Products, Guangdong Province Key Laboratory of Pharmacodynamic Constituents of TCM and New Drugs Research, College of Pharmacy, Jinan University, Guangzhou 510632, China
Hepatocellular carcinoma (HCC) is the fourth most common cause of cancer-related death worldwide as a severe global problem of public health, with over 80% of HCC cases occurring in low- and middle-resource countries, where medical and social care resources are often constrained [1]. As the main risk factor of HCC, chronic HBV infection accounts for most of global HCC cases of 257 million, especially in Asia-Pacific regions [2]. According to the latest statistics, on average, the disease progression for ~25% of untreated individuals will finally die of HCC and/or cirrhosis complications, and this rate even increases to 50% if only men are taken into consideration [3]. Currently, approved treatment agents for HCC are broadly classified into immunomodulatory agents [4, 5] and antiviral agents [6], the former including conventional IFNα-2b and Peg-IFNα-2a and the latter including nucleoside- or nucleotide-based reverse-transcriptase inhibitors such as nucleotide analogues (NUCs), nam replaced with other electroely, lamivudine, adefovir dipivoxil, entecavir, telbivudine, tenofovir disoproxil fumarate (TDF) and tenofovir alafenamide (TAF). The mainstay treatment for HCC in most countries is NUCs. Nevertheless, the clinical utilization of IFNα-2b and Peg-IFNα-2a is subject to a low response rates (about 20%–30%) among patients with chronic hepatitis B and a host of side effects, including flu-like symptoms, anemia, leucopenia, thrombocytopenia, anorexia, and depression [7]. Besides, the emergence of drug resistance during long-term NUCs-treatment is almost inevitable and represents a clinical challenge. For instance, approximately 70% of patients resistant to lamivudine [8] and entecavir also occurs among 43% lamivudine-pretreated patients after 4 years [9]. Accordingly, to improve the treatment of HCC and reduce drug resistance, there stands an urgency to develop novel agents with different therapeutic targets.
In recent years, targeted drugs for various types of cancer have developed rapidly [10-13]. In particular, the researches about drug development for HCC are increasing and attract a lot of interests [14, 15]. Sodium taurocholate cotransporting polypeptide (NTCP) is an integral membrane glycoprotein with seven predicted trans-membrane domains, which in humans is encoded by the SLC10A1 (solute carrier family 10 member 1) gene [16], and is expressed in the basolateral membranes of hepatocytes. Functionally, as a key component of the enterohepatic recovery of bile acids, NTCP participates in the enterohepatic circulation of bile acids. It is responsible for the basolateral uptake of bile acids from the portal blood into hepatocytes. What is more, NTCP was identified to serve as the functional receptor for HBV and HDV [17]. NTCP can specifically interact with the receptor-binding region of the pre-S1 domain of HBV/HDV envelope protein and thus virus gain access to cells. And further mechanism research has elucidated that NTCP can upregulate HBV transcription via farnesoid X receptor α (FxRα)-mediated activation of the HBV EN2/core promoter at the postentry step in an NTCP-dependent manner, and NTCP-targeting entry inhibitor manage to suppress HBV infection and replication [18]. Increasing researches have implicated that NTCP is involved in the progress of HCC: NTCP was down-regulated in HCC tissues and was up-regulated when HCC cell lines were arrested in the G0/ G1 phase [19]. Decreased SLC10A1/NTCP expression was also reported in a recent research about integrated proteogenomic characterization of HBV-related HCC [20]. Therefore, these findings have showed that NTCP inhibitors may offer a promising novel therapeutic option for HCC.
However, since homo NTCP was cloned 24 years ago, only a few human NTCP inhibitors have been identified [21, 22]. To date, the only recognized NTCP inhibitor is Myrcludex B (MyrB, phase Ⅱ of clinical trials had been conducted), a myristoylated synthetic lipopeptide comprising 47 amino acids derived from the pre-S1 domain of the HBV large surface protein. In the past few years, several FDA-approved drugs (Fig. 1A) were identified as NTCP inhibitors via a combination of computational and in vitro approaches [23], such as Fluvastatin [24], Macitentan [25], Bosentan [26] and Irbesartan [27]. Nevertheless, most of them have problems with target selectivity and side effects. For instance, because of inhibition of the uptake transporters NTCP and OATP/ Oatp, Fasiglifam may affect bile acid and bilirubin homeostasis, causing hyperbilirubinemia and cholestatic hepatotoxicity [28]. Besides, another posed challenge is that the full-size 3D crystal structure of NTCP is still not available, as a result, few NTCP inhibitors relied on reasonable structure-based de novo drug design are reported. Therefore, the objective of the present study is to develop a novel NTCP inhibitor. Herein, we eventually discovered a novel NTCP inhibitor B7 that exhibited good antiproliferative activity and induced apoptosis in HepG2 cells. This work reveals the potential of B7 as an NTCP inhibitor for future treatment of HCC.

|
Download:
|
| Fig. 1. (A) Several FDA-approved drugs identified as NTCP inhibitors. (B) The workflow of the virtual screening protocol for NTCP inhibitors. | |
Given the challenges in pharmacologically inhibiting NTCP, we developed a hybrid strategy combining computational and experimental approaches to screen potential NTCP inhibitors (Fig. 1B), homology modeling (Fig. S1 in Supporting information) was carried out to obtain the model structure of NTCP for followed screening. Next, the active site of inhibition is predicted and defined as the space where the helical structures of amino acids 84–87, 157–165 and 267 are embedded in view of the previous functional studies of NTCP from different species [29]. Then, we virtually docked more than 150, 000 compounds from the ZINC library by using LibDock based on the predicted site pocket of NTCP according to Lipinski's Rule of Five. Subsequently, the top 200 hits were further screened using the CDOCKER protocol. Resultant 10 hits with better CDOCKER interaction energies and LibDock scores were selected and purchased (Fig. S2 in Supporting information) on the basis of the top-ranked NTCP-compound binding models. Among these 10 hits, AK-968/12572207 showed the best antiproliferative activity with an IC50 of 22.913 ± 0.3 μmol/L as the leading compound for further structural modifications.
Next, according to molecular modeling, we analyzed the hydrophobicity of amino acid residues around the AK-968/12572207-NTCP binding pocket. We synthesized a series of analogs, A1–A24, containing different terminal substituent on the phenyl rings of benzamides or benzene sulfonamides on both sides (Scheme 1A) in order to fit the hydrophobic cavity at the side chain of Y108 and obtain suitable substitution vectors for improving activity. It was found that the anti-proliferation ability was improved when 4-position substituent of the phenyl rings was cyano or trifluoromethyl (IC50 = 20.922 ± 0.8 μmol/L and 14.970 ± 0.5 μmol/L), and both are electron-withdrawing groups. Intriguingly, while 4-position substituent group was replaced with other electron-with-drawing groups such as fluorine atom and trifluoromethoxy, the anti-proliferation ability was lower than that of the lead compound, which might suggest that the anti-proliferation ability was not completely determined by the electrophilicity of substituent groups. Meanwhile, compound A14 exhibited better anti-HBV activities than A6 and was therefore chosen for next optimization. Besides, we furtherly noticed the atmosphere enriched with a negative charge around N119 (Fig. S3 in Supporting information). Accordingly, we hypothesized that introduction of an appropriate positive charged group at the side chain of N119 may enhance the binding affinity. Therefore, B1–B8 with different positive charged groups were synthesized (Scheme 1B). Indeed, we obtained B2 and B7 through introduction of dibutylamine and 1-bocpiperazine, respectively, which exhibited better anti-proliferation ability with IC50 values of 10.881 ± 0.2 μmol/L and 7.360 ± 0.3 μmol/L, respectively (Table 1). Piperazinyl is a synergistic unit, which often appear in structures of anti-cancer drugs [30]. This may explain the excellent anti proliferation ability of B7. However, the other compounds of B series did not return increased feedback of the anti-proliferation ability, though the same positive charged groups were introduced, such as B4 and B5. We speculated that the accuracy of the homology modeling of NTCP was responsible for the difference. On the other hand, the anti-HBV activities of B series compounds were stronger than those of A series compounds, which may owe to the unique skeleton structure. Also, B2 and B7 also exhibited moderate anti-HBV activities, compared with Tenofovir as the positive antiviral control.

|
Download:
|
| Scheme 1. General procedures for the synthesis of compounds A1-A24 (A) and B1-B8 (B). Reagents and conditions: (a) Corresponding substituted benzenesulfonyl chloride or benzoyl chloride, CH2Cl2, -5 ℃, 1 h; (b) Corresponding substituted benzenesulfonyl chloride, CH2Cl2, -5 ℃, 1 h; (c) Chloroacetic chloride, CH2Cl2, -5 ℃, 1 h; (d) NHR2, CH3CN, 80 ℃, reflux, 6 h. | |
|
|
Table 1 Anti-proliferative activities and anti-HBV activities of the compounds.a |
{kind=link}
{kind=link}
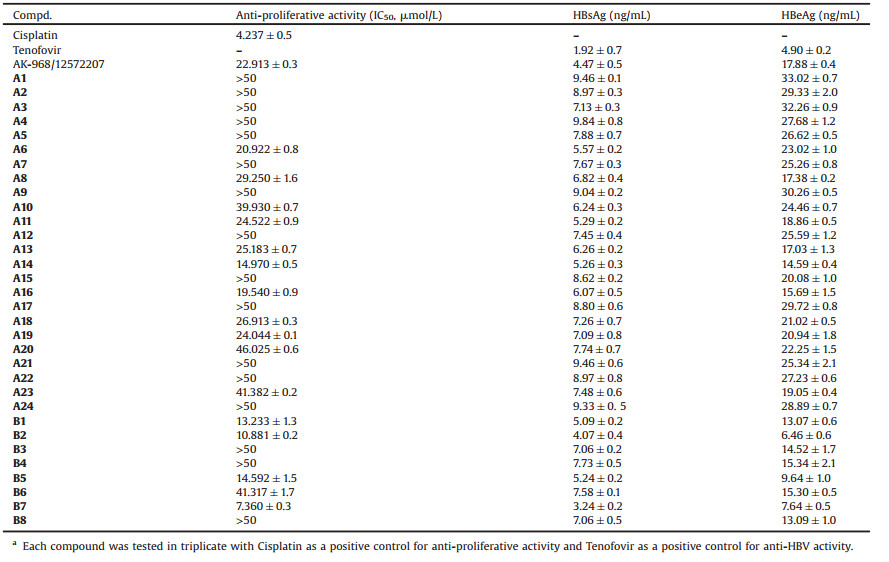
Cellular thermal shift assay (CETSA) has been developed as a popular method to monitor and quantify the extent to which a drug candidate reaches and directly binds to a protein target of interest within a cell since several years ago [31]. And as a result, plenty of researches have been utilizing CETSA for target verification [32]. And CETSA assessment by western blotting has revealed an increase in the melting temperature of NTCP (Figs. 2B and C), which confirmed the interaction of B7 (Fig. 2A) and NTCP. In addition, to demonstrate the stability of B7/NTCP complex, we also performed a 100 ns MD simulation on the B7/NTCP complex. The low root-mean-square deviation (RMSD) fluctuations and the convergence of the energies, temperatures, and pressures of the system indicated that it was a stable system (Fig. S4 in Supporting information). Furtherly, to explore the binding mode, the ligand structure with the most favorable binding free energies and reasonable orientations was selected as the optimal docked conformation, the result showed that the binding stability was mainly determined by hydrogen bonds (Fig. 2D). Thus, these results indicated that B7 was an NTCP inhibitor.

|
Download:
|
| Fig. 2. B7 binds to NTCP. (A) Chemical structure of B7. (B) CETSA assessment by western blotting. (C) Statistics of NTCP/β-actin. (D) Cartoon representation of NTCP model with docked B7. | |
{kind=link}
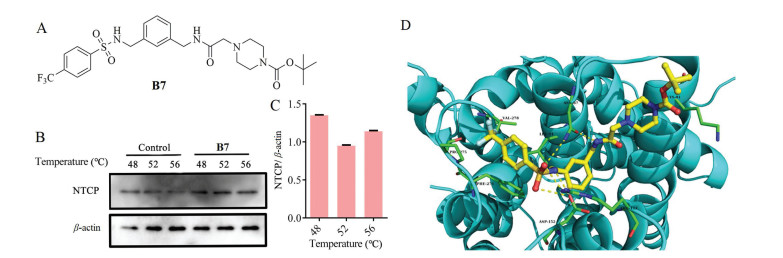
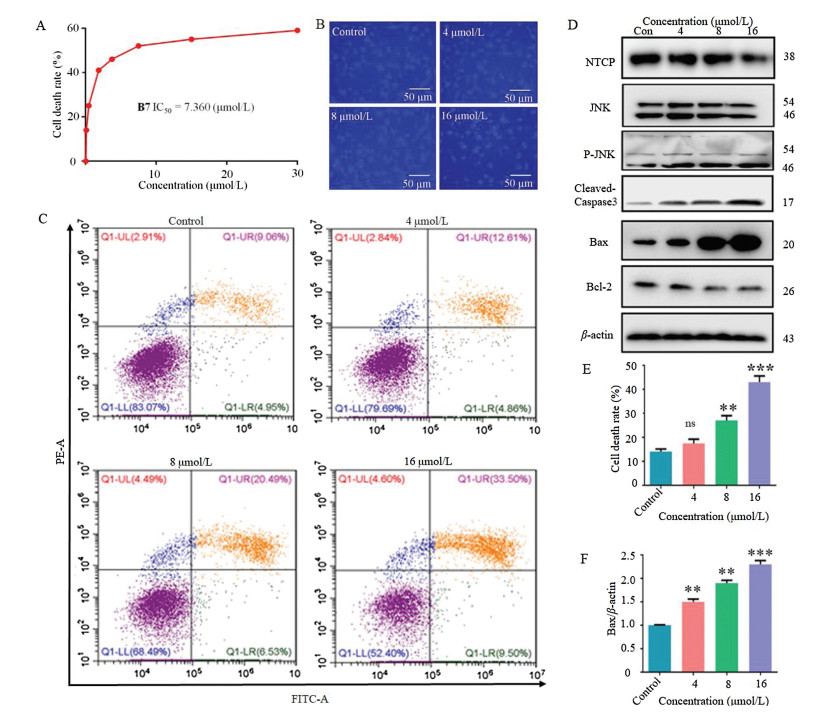
Compound B7 exhibited a favorable anti-proliferation effect on HepG2 cells and no significant toxicity in normal liver cells (Fig. S5A in Supporting information). Furtherly, to detect the effect of B7 on HCC cells, we firstly determined its effect on cell proliferation. B7 showed a robust dose-dependent anti-proliferation effect on HepG2 cells after 24 h (Fig. 3A). Next, we found a significant increase of apoptosis after B7 treatment in HepG2 cells. Condensed chromatin with bright fluorescence was observed in HepG2 cells after B7 treatment with Hoechst 33, 258 staining Fig. 3B). Additionally, Annexin-V/PI double staining also confirmed that B7 induced dose-dependent apoptosis (Figs. 3C and E). Next, we evaluated the JNK regulated classic mitochondrial apoptosis pathway and found B7 induced obviously up-regulation of JNK, p-JNK, caspase-3, Bax (Fig. 3F), and down-regulation of Bcl-2. Moreover, B7 could decrease the expression of NTCP which contributes to the progression of HCC (Fig. 3D). These results demonstrated that B7 exhibited an anti-proliferation effect on HepG2 cells by inducing apoptosis.

|
Download:
|
| Fig. 3. B7 induced apoptosis in HepG2 cells. (A) The cell inhibition rates were measured after being treated with different concentrations of B7 for 24 h. B Morphologic changes were observed under fluorescence microscopy with Hoechst 33, 258 staining after 24 h exposure to different concentrations of B7. Scale bar = 50 μm. (C) The apoptosis ratios were determined by flow cytometry analysis with annexin-V/PI double staining after 24 h exposure to different concentrations of B7. ns: not significance; ** P < 0.01, *** P < 0.001 vs. control. (D) The expressions of NTCP, JNK, p-JNK, caspase-3, Bax, and Bcl-2 were detected by western blot analysis after different concentrations of B7 treatment for 24 h. β-actin was used as the loading control. ** P < 0.01, *** P < 0.001 vs. control. (E) The cell death rate was measured after treated with different concentrations of B7. ns: not significance; ** P < 0.01, *** P < 0.001 vs. control. (F) Ratio of Bax/β-actin after cells were treated with different concentrations of B7. ns: not significance; ** P < 0.01, *** P < 0.001 vs. control. | |
{kind=link}
HCC induced by HBV infection remains one of the most prevalent yet undertreated public health problems. Furthermore, given the unmet therapeutic drug diversity and the emergence of drug-resistant of present agents, developing new small-molecules with novel targets represents an opportunity in HCC therapies, and thus, NTCP inhibitors may offer an alternative option for HCC treatment after taking the relationship between NTCP and HCC into consideration.
In summary, combining in silico high-throughput screening, chemical synthesis and anti-proliferative activity screening, we discovered a novel NTCP inhibitor B7 with favorable anti-proliferative activity against HepG2 cells. Moreover, we demonstrated that B7 could bind to NTCP and trigger apoptosis. These results could provide recommendable guidelines for the future development of NTCP-targeting small molecules and the treatment of HCC.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2018ZX09735005), the National Natural Science Foundation of China (Nos. 81922064, 81874290, 81673290, 81803347 and 81903502) and the Natural Science Foundation of Guangdong Province (No. 2018A030313707). Post-Doctor Research Project, West China Hospital, Sichuan University (No. 2019HXBH034).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.03.017.
| [1] |
J.D. Yang, P. Hainaut, G.J. Gores, et al., Nat. Rev. Gastro.Hepat. 16 (2019) 589-604. DOI:10.1038/s41575-019-0186-y |
| [2] |
H.B. El-Serag, Gastroenterology 142 (2012) 1264-1273. DOI:10.1053/j.gastro.2011.12.061 |
| [3] |
M.F. Yuen, D.S. Chen, G.M. Dusheiko, et al., Nat. Rev. Dis. Primers 4 (2018) 18035. DOI:10.1038/nrdp.2018.35 |
| [4] |
A. Jindal, A.K. Vyas, D. Kumar, G. Kumar, S.K. Sarin, Hepatol.Res. 48 (2018) 451-458. DOI:10.1111/hepr.13049 |
| [5] |
Z. Zhang, P. He, Y. Zhou, et al., Exp. Ther. Med. 16 (2018) 517-522. |
| [6] |
E.J. Gane, Liver Int. 37 (2017) 40-44. DOI:10.1111/liv.13304 |
| [7] |
R. Perrillo, Hepatology 49 (2009) S103-S111. DOI:10.1002/hep.22956 |
| [8] |
G.A. Tipples, M.M. Ma, K.P. Fischer, et al., Hepatology 24 (1996) 714-717. |
| [9] |
G.V. Papatheodoridis, S. Manolakopoulos, G. Dusheiko, A.J. Archimandritis, Lancet Infect. Dis. 8 (2008) 167-178. DOI:10.1016/S1473-3099(07)70264-5 |
| [10] |
L.Z. Ma, Z.Y. Zhang, S.P. Yin, et al., Chin. Chem. Lett. 30 (2019) 1942-1946. DOI:10.1016/j.cclet.2019.07.043 |
| [11] |
J.A. Zhao, H.B. Yu, S.C. Zhi, et al., Chin. Chem. Lett. 28 (2017) 1539-1546. DOI:10.1016/j.cclet.2017.03.025 |
| [12] |
Q. Xun, Z. Zhang, J. Luo, et al., J. Med. Chem. 61 (2018) 2353-2371. |
| [13] |
B. Yu, H. Liu, X. Kong, X. Chen, C. Wu, Eur. J. Med. Chem. 163 (2019) 500-511. DOI:10.1016/j.ejmech.2018.12.014 |
| [14] |
J.H. Xie, Y. Lu, B.Q. Yu, J. Wu, J. Liu, Chin. Chem. Lett. 31 (2020) 1173-1177. DOI:10.1016/j.cclet.2019.10.030 |
| [15] |
L. Chen, W. Qiu, J. Tang, Z.F. Wang, S.Y. He, Chin. Chem. Lett. 22 (2011) 413-416. DOI:10.1016/j.cclet.2010.10.036 |
| [16] |
B. Hagenbuch, P.J. Meier, J. Clin. Invest. 93 (1994) 1326-1331. DOI:10.1172/JCI117091 |
| [17] |
H. Yan, G. Zhong, G. Xu, et al., eLife 1 (2012) e00049. DOI:10.7554/eLife.00049 |
| [18] |
K. Zhao, S. Liu, Y. Chen, et al., Emerg. Microbes Infec. 7 (2018) 1-14. |
| [19] |
Y. Yan, L. Allweiss, D. Yang, et al., Emerg. Microbes Infec. 8 (2019) 879-894. DOI:10.1080/22221751.2019.1625728 |
| [20] |
Q. Gao, H. Zhu, L. Dong, et al., Cell 179 (2019) 561-577. DOI:10.1016/j.cell.2019.08.052 |
| [21] |
S.M. Zhao, Y.Q. Zhen, L.L. Fu, et al., Bioorg. Med. Chem. Lett. 29 (2019) 126623. DOI:10.1016/j.bmcl.2019.126623 |
| [22] |
J. Zhang, L.L. Fu, M. Tian, et al., Bioorg. Med. Chem. 23 (2015) 976-984. DOI:10.1016/j.bmc.2015.01.020 |
| [23] |
Z. Dong, S. Ekins, J.E. Polli, Mol. Pharmaceut. 10 (2013) 1008-1019. DOI:10.1021/mp300453k |
| [24] |
J.M. Donkers, B. Zehnder, G.J.P. van Westen, et al., Sci. Rep. 7 (2017) 15307. DOI:10.1038/s41598-017-15338-0 |
| [25] |
R. Greupink, L. Dillen, M. Monshouwer, M.T. Huisman, F.G.M. Russel, Eur. J. Pharm. Sci. 44 (2011) 487-496. DOI:10.1016/j.ejps.2011.09.009 |
| [26] |
E.I. Lepist, H. Gillies, W. Smith, et al., PLoS One 9 (2014) e87548. DOI:10.1371/journal.pone.0087548 |
| [27] |
E.M. Leslie, P.B. Watkins, R.B. Kim, K.L.R. Brouwer, J. Pharmacol. Exp. Ther. 321 (2007) 1170-1178. DOI:10.1124/jpet.106.119073 |
| [28] |
X.J. Wang, W. Hu, T.Y. Zhang, et al., Antivir. Res. 120 (2015) 140-146. DOI:10.1016/j.antiviral.2015.06.007 |
| [29] |
W. He, B. Ren, F. Mao, et al., PLoS Pathog. 11 (2015) e1004840. DOI:10.1371/journal.ppat.1004840 |
| [30] |
X.L. Chen, H.W. Jia, Z.Y. Li, X. Xu, Chin. Chem. Lett. 30 (2019) 1207-1213. DOI:10.1016/j.cclet.2019.02.033 |
| [31] |
R. Jafari, H. Almqvist, H. Axelsson, et al., Nat. Protoc. 9 (2014) 2100-2122. DOI:10.1038/nprot.2014.138 |
| [32] |
L. Laraia, A. Friese, D. Corkery, et al., Nat. Chem. Biol. 15 (2019) 710-720. DOI:10.1038/s41589-019-0307-5 |