 2020, Vol. 31
2020, Vol. 31 


To meet the increasingly stringent regulations in China, largescale of control devices have been implemented at the source of production to reduce the emission of air pollutants. However, in the operation of these devices, secondary pollution occurs frequently, which makes many of the mature technologies appear to be challenged [1-3]. Therefore, exploration of the secondary pollution and control strategies in the process of air pollution control have practical significance for assessing the environmental risk and improving the current technologies.
As one of national priority control pollutants, chlorinated organics are inclined to transfer into polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs, also referred as dioxins) in the catalytic elimination process, which has been a grave challenge in environmental catalysis [4-6]. The PCDD/Fs are listed as Persistent Organic Pollutants (POP) by the Stockholm Convention. These compounds have attracted much social and scientific attention as they are among the most toxic chemicals on the earth. The vast majority of PCDD/Fs in the environment originate from anthropogenic sources, which include municipal solid waste incineration (MSWI), iron ore sintering (IOS), and metal smelting and coking [7-9]. So far there is no statistics released by China showing the national annual emission of PCDD/Fs. However, the Chinese emission standard for PCDD/Fs (e.g., 0.1 ng I-TEQ Nm-3 for MSWI) is the same as EU standard, putting considerable pressure on many industrial thermal-treatment processes.
Apart from the aforementioned sources, the formation of PCDD/Fs from chlorinated organics elimination process should not be negligible. Recent report shows that in pharmaceutical industries (PIs), PCDD/Fs ranging from 0.148 ng/m3 to 9.051 ng/m3 and 0.008 to 0.255 ng I-TEQ Nm-3 were detected in the off-gases of 16 regenerative thermal oxidizer (RTO) devices [10]. Although the emission levels of most RTOs are lower than the national standard, annual emission of 19.96 g I-TEQ PCDD/Fs has been estimated owning to the large amounts of PIs in China. Furthermore, recent laboratory tests confirmed that the PCDD/Fs could be generated in the catalytic oxidation of 1, 2-dichlorobenzene (DCBz) over CuOx/ TiO2-CNTs [11] and V2O5-WO3/TiO2 catalysts [12]. Our work further revealed that the conversion of chlorobenzene (CBz) into PCDD/Fs could also occur in a synergistic selective catalytic reduction (SCR) reaction [5]. Since mg/m3 level of CBz have been detected in coal combustion process [13], and most of power plants and industrial boilers have installed the SCR system, secondary pollution generated from these combustion sources should be also taken concern.
In the past decades, a large number of catalyst systems have been explored for the efficient elimination of chlorinated organics [4, 14, 15]. However, most of the works focused on how to improve the destruction efficiencies of applied catalysts, while few efforts are devoted to explore the potential formation of PCDD/Fs in the catalytic process. Liu et al. recently studied the distributions of polychlorinated benzenes (PCBz) in the catalytic oxidation of CBz over a range of noble metals (e.g., Pd, Pt, Ru, Rh) [16], but did not reveal insights into the PCDD/F formation. In this work, active species including Pt, Ru, Mn, Ce and V were loaded onto carbon nanotubes (CNTs). The support was employed to capture the potentially generated PCDD/Fs in the process of catalytic CBz oxidation (CBCO). Samples were subjected to a feed stream containing 500 ppm of CB, 10 vol% O2, and balanced N2 at a WHSV = 20, 000 mL g-1 h-1. As reported by Ji et al. [12], there was no major difference in the homologue profiles of PCDD/Fs between 11 vol% and 2 vol% O2, but the PCDD/F-output escalated rapidly with decreasing the oxygen content. Accordingly, a sufficiently high 10 vol% O2 was applied herein for heath safety consideration of laboratory tests. Furthermore, the generation of PCDD/Fs from a heterogeneous pathway generally proceeds between 200 ℃ and 400 ℃ [17, 18]. As such, a moderate ageing temperature of 250 ℃ was selected. Details of reaction measurements, byproducts analyses and catalyst characterizations were provided in Supporting information.
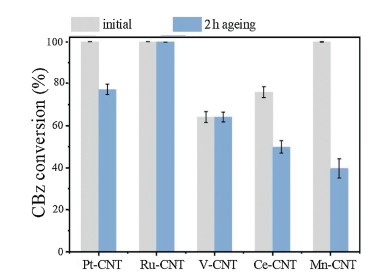
As shown in Fig. 1, the Ru-CNT and V-CNT samples did not show apparent deactivation after a relatively short-term ageing, while other samples exhibited a significant decrease in CBz conversion efficiency. The deactivation of noble metal- and transition metal oxide-based catalysts has been shown to mainly originate from Cl poisoning [14], where the chlorine accumulation on the catalyst active sites resulted in the inhibition of CBz oxidation, and could meanwhile induce an electrophilic chlorination that generated moretoxic polychlorinated byproducts. Indeed, after measuring the reaction byproducts in the off-gases, all of the samples were found to produce significant byproducts. As shown in Fig. 2 and Table 1, these byproducts mainly involved chlorinated alkanes such as trichloromethane (CHCl3), carbon tetrachloride (CCl4), trichloroethylene (C2HCl3) and tetrachloroethylene (C2Cl4). In particular, the PCBz including dichlorobenzene (C6H4Cl2) and trichlorobenzene (C6H3Cl3) were also detected. Further extracting the used catalysts by using the dichloromethane, these PCBz were also observed on the catalyst surface, together with some chlorophenols (CPs) (Fig. S1 in Supporting information). It is well known that the PCBz are more toxic than CBz, which could be further transferred into PCDD/Fs via a series of heterogeneous reactions (e.g., Ullmann reaction) [18, 19], and the CPs are considered as the main precursor for PCDD/F formation. As such, the observation of PCBz and CPs revealed an environmental risk for applying these active species in the catalytic elimination of chlorinated organics. Additionally, although the Ru-CNT and V-CNT samples revealed no deactivation after ageing, they still produced many chlorinated byproducts during the CBCO reaction.

|
Download:
|
| Fig. 1. CBz conversion efficiency at 250 ℃ with the two periods of ageing time. Reaction condition: 500 ppm CB, 10 vol% O2, N2 balance and WHSV 20, 000 mL g-1 h-1. | |

|
Download:
|
| Fig. 2. GC/MS qualitative analyses of gaseous products generated from the CBCO reaction at 250 ℃ with two periods of ageing time. Reaction condition: 500 ppm CB, 10 vol% O2, N2 balance and WHSV 20, 000 mL g-1 h-1. | |
|
|
Table 1 GC/MS qualitative analyses of gaseous products generated from the CBCO reaction at 250 ℃. |

{kind=link}
{kind=link}
Among these samples, the Pt-CNT was shown to yield the most dichlorobenzene and trichlorobenzene in the off-gas. The formation of oxychlorides species (PtOxCly) in the presence of sufficient O2 has been reported being responsible for this [20-22]. The origin of PCBz in the Mn-CNT and Ce-CNT samples has been demonstrated to be caused by the attack of Cl (that were captured at oxygen vacancies) with adsorbed CBz [23, 24], where the Mn-CNT was found to yield much higher PCBz than the Ce-CNT, suggesting a more severe electrophilic chlorination in the sample. The Ru-CNT likely proceeded a semi-Deacon reaction (2Cl- + O2 + V0 = Cl2 + 2Olat) in the CBCO reaction [15, 25, 26]; this efficiently desorbed the accumulated Cl from catalyst surface, and resulted in robust CB oxidation (Fig. 1) and less PCBz/CPs generation. The VOx has shown with Brønsted and Lewis acidities, which originate from its V-OH and V=O groups, respectively [27, 28]. The presence of Brønsted V-OH could act as proton source, which reacted with Cl forming HCl [29, 30], facilitating the Cl desorption and inhibiting the electrophilic chlorination.
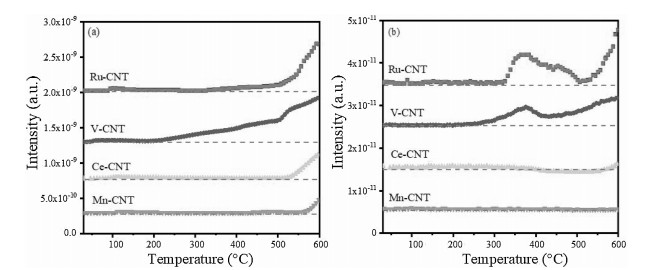
To further verify aforementioned Cl reaction and desorption behaviors, temperature-programmed surface reaction (TPSR) measurements were conducted by purging the CBz and O2 onto each sample and monitored their reaction products using a Mass spectroscopy (MS). As shown in Fig. 3, HCl appeared at approximately 200 ℃ in the V-CNT sample, the amount of which was distinctly increased with elevating temperature. This result confirmed the assumption that the V-OH groups provided the H proton to facilitate HCl formation. The Ru-CNT exhibited an intense Cl2 desorption peak initiating at the temperature of approximately 300 ℃, indicating that a semi-Deacon reaction indeed occurred. For the Ce-CNT and Mn-CNT, no significant desorption was observed for both HCl and Cl2 at the temperatures below 500 ℃. This suggested that rather poor Cl desorption appeared in these two samples. In general, the cleavage of C-Cl was an initial step for CBCO reaction as the C-Cl bond energy (at 339 kJ/mol) was much lower than that of C-H (414 kJ/mol). At Lewis CeO2 and MnOx surface, such a cleavage process preferred to occur at oxygen vacancy, followed by a nucleophilic attack with CBz [31]. This dissociated the Cl at the surface oxygen vacancy and formed phenyls attached to the surface lattice oxygen atoms. The phenyl groups were then attacked by H (from the surface hydroxyls) forming the benzene (see label 7 of benzene in the Fig. 2) [23]. In the Ce-CNT and Mn-CNT, the captured Cl were very stable, which led to rapid deactivation of the samples (Fig. 1), and promoted the electrophilic chlorination, increasing the generation of polychlorinated byproducts. In comparison with Mn-CNT, Ce-CNT yielded much less polychlorinated byproducts. This could be attributed to the limited surface chlorination of CeO2 as verified by our previous density functional theory (DFT) calculation [31], which indicated, as the temperature increases, the replacement of chlorine species located in the prechlorinated oxide surface by oxygen becomes much more thermodynamically feasible, and the prechlorination of the surfaces tends to promote the resistance against higher concentration of Cl2 in the equilibrium gaseous phase. Further X-ray photoelectron spectroscopy (XPS) analyses also confirmed the presence of much less intense Cl 2p characteristic bands on the aged Ce-CNT surface (Fig. S2 in Supporting information) [32].

|
Download:
|
| Fig. 3. CBz-TPSR profiles of (a)HCl and (b)Cl2 for Ru-CNT, V-CNT, Ce-CNTand Mn-CNTcatalysts. Reaction condition: 500 ppm CB, 10 vol% O2, N2 balance, WHSV20, 000 mL g-1 h-1. | |
{kind=link}
To evaluate the potential generation of PCDD/Fs in the catalytic elimination process, the off-gases of CBCO reaction were collected by absorption in a 100 mL toluene, and the used catalysts were extracted, both of which were subsequently analyzed using a HR-GC/HR-MS system following a standard PCDD/F measurement procedure. Since the Pt-CNT sample exhibited remarkably high PCBz byproducts, only Ru was selected as a representative noble metal for further analysis in consideration of the health safety issue. In general, PCDDs have 75 congeners, among which 7 congeners have high toxicity, and PCDFs have 135 congeners, 10 of which have high toxicity. Therefore, the 17 congeners were measured herein for comparison.
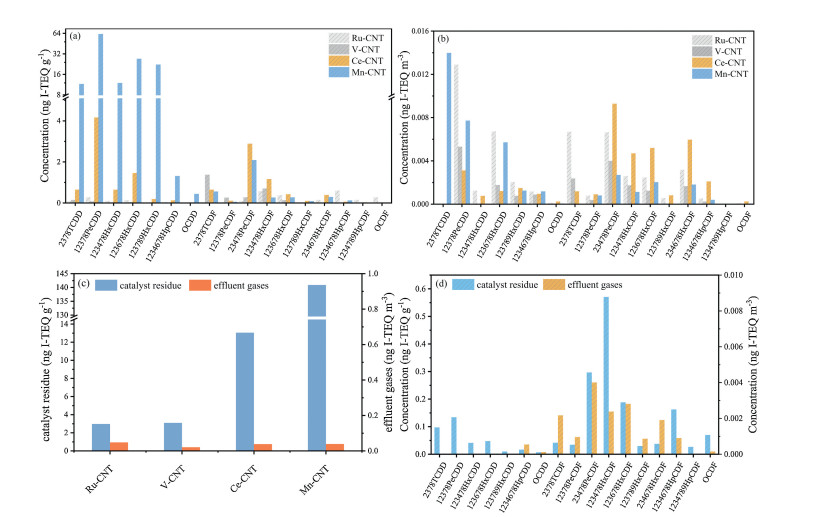
As shown in Figs. 4a and b, the majority of PCDD/Fs have been captured by CNTs as only few of them were detected in the offgases. After ageing at 250 ℃ for 30 h, the most toxic 2, 3, 7, 8-TCDD was observed in the Ce-CNT and Mn-CNT, which were rarely generated in the Ru-CNT and V-CNT. The sequence of total concentration of PCDD/Fs was Mn-CNT > Ce-CNT > Ru-CNT ≥ V-CNT (Fig. 4c), while the Mn-CNT yielded the most PCDD/Fs with the total concentration at 0.039 ng I-TEQ m-3 in the effluent and 140.9 ng I-TEQ g-1 on the catalyst surface. The homologue profile suggested that Mn-CNT was with the primary PCDDs, which has been reported originating from CPs [33-35] (Fig. S1). It is noteworthy that the total concentration of PCDD/Fs captured by CNTs was remarkably higher than Chinese national standard of solid residue (at 3 ng I-TEQ g-1) in the Mn-CNT, so did the Ce-CNT that had the value of 13.1 ng I-TEQ g-1. Furthermore, the 17 toxic PCDD/F congeners were all detected in the four samples. Both of these provided direct evidence that secondary pollution indeed occurred in the catalytic elimination of chlorinated organics, and seems to be universal for all industrially applied active species. Additionally, even we used dichloromethane (DCM) as reaction precursor, significant dioxins were still detected in the effluent and on the catalyst surface for Mn-CNT (Fig. 4d). This is the first report of DCM conversion to PCDD/Fs under laboratory tests, which is in line with the observation of PCDD/Fs from the 16 RTOs in PIs as the DCM is a major chlorinated pollutant at this source. It is likely that the DCM might initially precede an aromatization process, following by a condensation reaction to form the dioxins. Further work should be conducted to explore the underlying mechanism for this important conversion process.

|
Download:
|
| Fig. 4. The homologue profiles of PCDD/Fs (a) on catalyst residue and (b) in the effluents after ageing at 250 ℃ for 30 h on each catalyst; (c) the total concentrations of PCDD/Fs for each catalyst; (d) the homologue profile of PCDD/Fs using the DCM as precursor on Mn-CNT catalyst. | |
{kind=link}
The formation of dioxins through either homogenous or heterogeneous pathway has shown to be very complex. In the past decades, significant efforts have been devoted to reveal the underlying mechanism of PCDD/F formation [19, 36]. Some consensuses have been reached but there are still many features largely unknown [17]. In this study, we only aim to provide direct evidences for the formation of PCDD/Fs in the process of catalytic elimination of chlorinated organic pollutants. We demonstrated that a range of industrially important catalyst systems, such as Ru, V, Ce and Mn species would all produce the PCDD/Fs in CBCO reaction. Amongst which, the Mn specie was shown to be the most active for PCDD/F formation, even with the DCM as precursor. After ageing at 250 ℃ for 30 h, the PCDD/Fs captured by CNTs on both the Mn and Ce species were remarkably higher than Chinese national standard, and TPSR measurements suggested that such a high amounts of PCDD/F generation could be strongly correlated with their insufficient desorption of Cl from the active sites. Although the chemical features of PCDD/Fs formation in the catalytic elimination process are not explored herein, detailed mechanism studies are currently conducted, the results of which will be reported in due course.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21777140, 21922607) and the Outstanding Youth Project of Zhejiang Natural Science Foundation (No. LR19E080004).
Appendix A. Supplementary dataSupplementary material related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.03.056.
| [1] |
W.L. Wang, Q. Meng, Y. Xue, et al., J. Catal. 366 (2018) 213-222. DOI:10.1016/j.jcat.2018.07.022 |
| [2] |
M. Zhu, L. Zhang, S. Liu, et al., Chin. Chem. Lett. (2020). DOI:10.1016/j.cclet.2020.01.017 |
| [3] |
J. Zou, Y. Chen, S. Liu, et al., Water Res. 150 (2019) 330-339. |
| [4] |
C. He, J. Cheng, X. Zhang, et al., Chem. Rev. 119 (2019) 4471-4568. DOI:10.1021/acs.chemrev.8b00408 |
| [5] |
W. Jiang, Y. Yu, F. Bi, P. Sun, X. Weng, Z. Wu, Environ. Sci. Technol. 53 (2019) 12657-12667. DOI:10.1021/acs.est.9b04155 |
| [6] |
X. Dai, X. Wang, Y. Long, et al., Environ. Sci. Technol. 53 (2019) 12697-12705. DOI:10.1021/acs.est.9b05088 |
| [7] |
Y. Fan, M. Ren, H. Zhang, et al., Chemosphere 224 (2019) 298-305. DOI:10.1016/j.chemosphere.2019.02.117 |
| [8] |
Y. Yang, G. Wu, C. Jiang, et al., Environ. Pollut. 260 (2020) 113946. DOI:10.1016/j.envpol.2020.113946 |
| [9] |
Z. Peng, R. Weber, Y. Ren, et al., Waste Manage. 103 (2020) 260-267. DOI:10.1016/j.wasman.2019.12.028 |
| [10] |
M. Zhan, Y. Ma, X. Lin, et al., Aerosol Air Qual. Res. 19 (2019) 2070-2082. DOI:10.4209/aaqr.2019.06.0284 |
| [11] |
Q. Wang, Q. Huang, H. Wu, et al., Chemosphere 144 (2016) 2343-2350. DOI:10.1016/j.chemosphere.2015.10.097 |
| [12] |
L. Ji, X. Cao, S. Lu, et al., J. Hazard. Mater. 342 (2018) 220-230. DOI:10.1016/j.jhazmat.2017.07.020 |
| [13] |
J. Cheng, Y. Zhang, T. Wang, et al., Fuel 225 (2018) 554-562. DOI:10.1016/j.fuel.2018.03.185 |
| [14] |
C. Du, S. Lu, Q. Wang, et al., Chem. Eng. J. 334 (2018) 519-544. DOI:10.1016/j.cej.2017.09.018 |
| [15] |
Q. Dai, J. Wu, W. Deng, J, et al., Appl. Catal. B 249 (2019) 9-18. DOI:10.1016/j.apcatb.2019.02.065 |
| [16] |
X. Liu, L. Chen, T. Zhu, R. Ning, J. Hazard. Mater. 363 (2019) 90-98. DOI:10.1016/j.jhazmat.2018.09.074 |
| [17] |
M. Altarawneh, B.Z. Dlugogorski, E.M. Kennedy, J.C. Mackie, Prog. Energy Combust. Sci. 35 (2009) 245-274. DOI:10.1016/j.pecs.2008.12.001 |
| [18] |
R. Addink, K. Olie, Environ. Sci. Technol. 29 (1995) 1425-1435. DOI:10.1021/es00006a002 |
| [19] |
B.R. Stanmore, Flame 136 (2004) 398-427. DOI:10.1016/j.combustflame.2003.11.004 |
| [20] |
R.W. van den Brink, R. Louw, P. Mulder, Appl. Catal. B 16 (1998) 219-226. DOI:10.1016/S0926-3373(97)00076-3 |
| [21] |
S. Scirè, Appl. Catal. B 45 (2003) 117-125. DOI:10.1016/S0926-3373(03)00122-X |
| [22] |
M. Taralunga, J. Mijoin, P. Magnoux, Appl. Catal. B 60 (2005) 163-171. DOI:10.1016/j.apcatb.2005.02.024 |
| [23] |
H.A. Miran, M. Altarawneh, Z. Jiang, et al., Catal. Sci. Technol. 7 (2017) 3902-3919. DOI:10.1039/C7CY01096F |
| [24] |
X. Weng, Y. Long, W. Wang, M. Shao, Z. Wu, Chin. J. Catal. 40 (2019) 638-646. DOI:10.1016/S1872-2067(19)63322-X |
| [25] |
Q. Dai, S. Bai, J. Wang, et al., Appl. Catal. B 142- 143 (2013) 222-233. |
| [26] |
Q. Dai, S. Bai, X. Wang, G. Lu, Appl. Catal. B 129 (2013) 580-588. DOI:10.1016/j.apcatb.2012.10.006 |
| [27] |
H. Huang, Y. Gu, J. Zhao, X. Wang, J. Catal. 326 (2015) 54-68. DOI:10.1016/j.jcat.2015.02.016 |
| [28] |
J. Wang, X. Wang, X. Liu, et al., J. Mol. Catal. A 402 (2015) 1-9. DOI:10.1016/j.molcata.2015.03.003 |
| [29] |
S. Albonetti, S. Blasioli, R. Bonelli, et al., Appl. Catal. A 341 (2008) 18-25. DOI:10.1016/j.apcata.2007.12.033 |
| [30] |
Q. Dai, L. Yin, S. Bai, et al., Appl. Catal. B 182 (2016) 598-610. DOI:10.1016/j.apcatb.2015.10.016 |
| [31] |
W. Cen, Y. Liu, Z. Wu, et al., J. Phys. Chem. C 118 (2014) 6758-6766. |
| [32] |
X. Wang, L. Ran, Y. Dai, Y. Lu, Q. Dai, J. Colloid Interface Sci. 426 (2014) 324-332. |
| [33] |
M.S. Milligan, E.R. Altwicker, Environ. Sci. Technol. 30 (1996) 225-229. DOI:10.1021/es9502583 |
| [34] |
J.G.P. Born, P. Mulder, R. Louw, Environ. Sci. Technol. 27 (1993) 1849-1863. DOI:10.1021/es00046a013 |
| [35] |
S. Nganai, B. Dellinger, S. Lomnicki, Environ. Sci. Technol. 48 (2014) 13864-13870. DOI:10.1021/es504253w |
| [36] |
S. Lomnicki, B. Dellinger, J. Phys. Chem. A 107 (2003) 4387-4395. DOI:10.1021/jp026045z |