 2020, Vol. 31
2020, Vol. 31 


Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) spreading through the respiratory tract [1], remains one of the leading causes of infectious disease. It has been estimated that in 2018, nearly 10 million people worldwide were infected with TB and 1. 4–1. 6 million people died from the disease [2]. The standard TB treatment recommended by World Health Organization (WHO) is generally comprised of treatment for 2 months in the intensive phase with isoniazid (INH), rifampicin (RIF), ethambutol (EMB) and pyrazinamide (PZA), followed by a further 4 months of isoniazid and rifampicin therapy in a continuation phase [3]. However, the emergence of multidrug-resistant tuberculosis (MDR-TB) and extensive drug-resistant tuberculosis (XDR-TB) strains has greatly reduced the clinical therapeutic management of TB. Prior to the approval of bedaquiline (1, Fig.1), MDR-TB was treated with a combination of five to seven drugs for up to 18–24 months and there was an urgent need to find novel drugs with the potential to accelerate the treatment or to address MDR-TB and XDR-TB specifically.

|
Download:
|
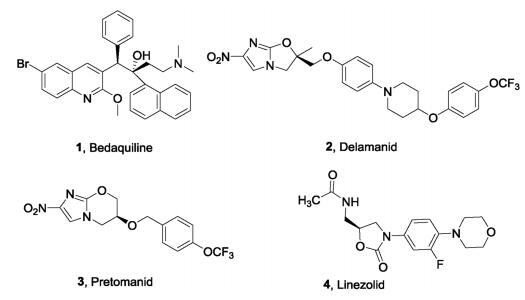
| Fig. 1. Antituberculosis drugs approved between 2012 and 2019. | |
{kind=link}
Bedaquiline (1, TMC-207), a diarylquinoline, was approved by the Food and Drug Administration (FDA) in 2012 as a component of the combination therapy for the treatment of adult patients affected by MDR-TB. Bedaquiline is effective not only for drugsensitive and drug-resistant tuberculosis, but also combats replication and dormant bacilli [4, 5]. Bedaquiline is the first approved antituberculosis drug in almost 40 years. It targets the energy-generating machinery of F1Fo-ATP synthase in Mtb, confirming that components of the oxidative phosphorylation pathway which supplies energy to Mtb can serve as effective targets for the treatment of TB, although bedaquiline has beenassigned a black box warning for safety risks [6]. In 2014, the nitro-2,3-dihydroimidazo[2,1-b]oxazolebased drug delamanid (2, OPC67683, Fig.1) was approved by the European Medicines Agency (EMA) for the treatment of MDR-TB [7]. Furthermore, FDA recently approved a combination of another nitroimidazole, pretomanid (3, Fig.1), with bedaquiline and the oxazolidinonebased linezolid (4, Fig.1) to treat adult patients with XDR-TB [8].
The emergence of MDR-TB and XDR-TB inspired scientists to search for novel targets for antituberculosis drug development, preferably with no cross-resistance with existing drugs. The mechanism of action of antimycobacterial drugs includes inhibition of cell wall biosynthesis, mycolic acid biosynthesis, protein synthesis, replication or transcription and energy production and metabolism [9, 10]. The recent discovery of small molecules targeting energy generation in Mtb, especially for the confirmed ATP synthase inhibitor bedaquiline (1), confirmed that the inhibition of the oxidative phosphorylation pathway for energy generation is an attractive strategy in Mtb. Other proteins that can effect energy generation in the respiratory chain have been explored as potential drug targets. These include non-proton-pumping type II NADH dehydrogenase (NDH-2) and the b subunit of cytochrome bcc-aa3 complex (QcrB), for which clinical drugs TBI-166 and Q203 were found to be inhibitory. In this review, we summarize the main compositions and electron transfer process of the mycobacterial respiratory chain, and the recent progress of new small molecule drugs targeting NADH-2, QcrB and ATP synthase, the main three components in the respiratory chain. We describe the optimization strategy of QcrB inhibitors obtained in our laboratory and discuss challenges of QcrB inhibitors as antituberculosis drugs as a result of the alternate cytochrome bd oxidase oxidative compensation pathway.
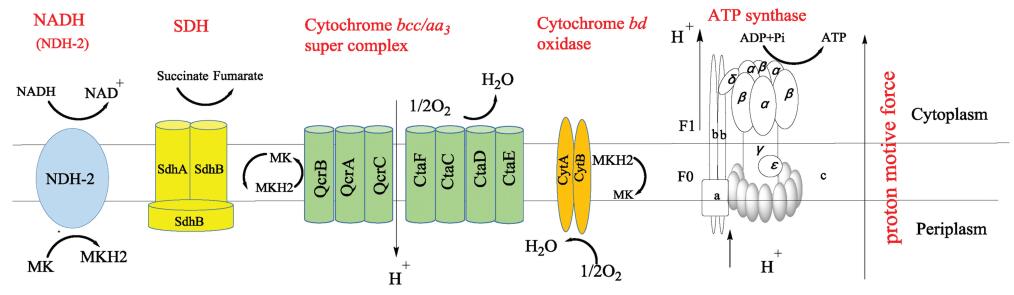
2. Respiratory chain of MtbBacterial processes usually gain energy by substrate level phosphorylation or by oxidative phosphorylation. Most processes gain energy using substrate level phosphorylation, but on the contrary, Mtb produces adenosine triphosphate (ATP) and maintains its vitality mainly through oxidative phosphorylation [11]. The energy produced in association with the metabolism of mycobacteria, especially in the oxidative phosphorylation pathway, has become an important target in antituberculosis drug discovery [12, 13]. As shown in Fig.2, electrons enter the electron transport chain via NADH dehydrogenase, resulting in reduction of the menaquinone (MK) pool. After accepting electrons from NDH-2, menaquinol can be re-oxidized by either of two enzymes, cytochrome bcc-aa3 complex or cytochrome bd type terminal oxidase. In the entire oxidative phosphorylation process, electrons are transferred between the membrane-bound primary dehydrogenases and the terminal oxidase, resulting in production of proton motive force (PMF), after which ATP synthase can produce ATP using the PMF generated from this process. Blocking the introduction of electrons into the respiratory chain can prevent the synthesis of ATP, and this has led to a search for drugs targeting the oxidative phosphorylation pathway in respiratory chain of Mtb [14].

|
Download:
|
| Fig. 2. Schematic view of the mycobacterial respiratory chain (MK: menaquinone, MKH2:menaquinol, NDH-2: type II NADH dehydrogenase, SDH: succinate dehydrogenase). | |
{kind=link}
3. Three important druggable targets in the respiratory chain 3.1. Proton-pumping type II NADH dehydrogenase (NDH-2)
Electrons are introduced into the electron transport chain (ETC) primarily via NADH dehydrogenases. Most bacteria contain three NADH dehydrogenases: (1) sodium-pumping NADH dehydrogenase (NQR), (2) proton-pumping type I NADH dehydrogenase (NDH-1), and (3) non-proton-pumping type II NADH dehydrogenase (NDH-2) [15]. Mtb has both NDH-1 and NDH-2 and uses themto harness the oxidation of NADH from central metabolism to energize the electron transport chain [16]. The difference between NDH-1 and NDH-2 is that NDH-1 is usually expressed under aerobic conditions, whereas NDH-2 is active under anaerobic conditions [17]. Previous studies have shown that rotenone, an NDH-1 inhibitor, could not kill hypoxia-adapted Mtb cells, which suggests that NDH-1 does not represent a compelling target for drug development [18]. NDH-2 on the other hand, a single subunit oxidoreductase with two copies (Ndh and NdhA) with 67% sequence identity [19], is essential for ATP synthesis, promoting the reduction of MK to MKH2 while oxidizing NADH to NAD+ [20]. Treatment of mycobacteria with an NDH-2 inhibitor, trifluoperazine leads to blockage of initiation of the ETC of mycobacteria under anaerobic conditions [18]. NDH-2 is not found in mammalian mitochondria and these features make NDH-2 a potential drug target for TB [21].
3.2. Cytochrome oxidaseIn the aerobic respiration of Mtb, there are two kinds of cytochrome oxidases which can transfer electrons to oxygen after menadione is reduced by the electrons of NDH-2. One is the supercomplex of menaquinol-cytochrome c oxidoreductase (cytochrome bcc/Qcr)-aa3 cytochrome c oxidase (CtaC-F) complex. The other is cytochrome bd type menaquinol oxidase which is encoded by cydABDC [22, 23]. Both branches can transfer electrons to generate oxygen from water, releasing energy, but they have different efficiencies. The bcc-aa3 branch pumps protons out of the cell during the transfer of electrons to oxygen in conditions of aerobic replication. In 2018, Gong et al. reported a cryo-EM crystal structure of the cytochrome bcc-aa3 supercomplex isolated from M. smegmatis [24]. This structure is comprised of a cytochrome bcc (complex III) dimer flanked on either side by aa3 cytochrome c oxidase (complex IV) subunits. The cytochrome bcc complex contains QcrA, QcrB and QcrC subunits (Fig.2), which transfer the electrons to CtaC-F. The QcrB subunit contains two binding sites, a quinol oxidation site (QP site) and a quinone reduction site (QN site). The QP site is responsible for the oxidation of menaquinol (MKH2) to menaquinone (MK) and is located close to heme bL, while the QN site is responsible for reduction of MK into MKH2 and is close to heme bH. High-throughput screening (HTS) has identified a series of imidazopyridinamide (IPA) compounds that interfere in this energy metabolism [25, 26]. A clinical candidate, Q203 targets the QcrB subunit of the cytochrome bcc complex by competing with MHK2 for binding at the QP site [24, 27, 28].
Compared with the cytochrome bcc-aa3 branch, cytochrome bd oxidase is less efficient because it does not pump protons but instead generates a PMF by transmembrane charge separation [29]. Cytochrome bd oxidase is an integral membrane protein complex and consists of two main subunits, CydA and CydB (Fig.2) [30]. It also plays an important role in resisting different types of stress, such as low oxygen tension or the presence of nitric oxide or peroxide [31]. Notably, studies have shown that when the cytochrome bcc complex is damaged by a deletion mutation, the cytochrome bd branch may compensate, at least partially, for the insufficient function of the cytochrome bcc-aa3 branch of the respiratory chain [32, 33]. However, there is no validated inhibitor of cytochrome bd oxidase to be reported.
3.3. F1Fo-ATP synthaseATP synthase is a ubiquitous enzyme, a key in the energy metabolismof virtually all cells, and is essential for the growth of mycobacteria on fermentable and non-fermentable carbon sources [11]. During the process of electron transfer from NADH or succinic acid to a terminal electron acceptor for Mtb energy metabolism, F1Fo-ATP synthase a macromolecular, membrane-embedded protein complex encoded by the ATPase BEFHAGDC operon, Rv1304-1311, can synthesize ATP by utilizing an electrochemical gradient of protons or sodium ions to catalyze the reaction of ADP with inorganic phosphate (Pi) [4, 34]. The X-ray structure of F1Fo-ATP synthase shows that it consists of a transmembrane Fo section (subunits a, b and c) with a cytosolic F1 segment comprising subunits α, β, γ, δ and ε that catalyzes ATP generation (Fig.2) [35, 36]. The Fo segment embedded in the membrane has a subunit composition of a1b2c8-15, while the hydrophilic F1 part consists of an α3β3γδε subunit (Fig.2) [37, 38]. An important step in proton transport is the binding of protons to the essential acidic residues in the central transmembrane portion of subunit c. Bedaquiline (1) targets the mycobacterium subunit c of F1Fo-ATP synthase leading to cellular ATP depletion and ultimately to the biological death of Mtb [39, 40]. The success of bedaquiline further confirms that ATP synthesis has become a key area in which new drug targets for mycobacteria can be identified.
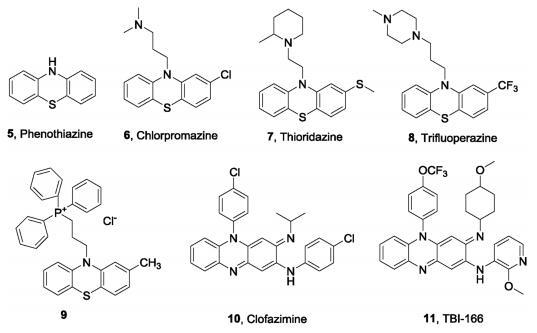
4. Medicinal chemistry efforts targeting the respiratory chain 4.1. NDH-2 inhibitorsPhenothiazine (5) (Fig.3), such as chlorpromazine (6), thioridazine (7), trifluoperazine (8), have been reported to have antimycobacterial activity against drug-susceptible and drug resistant Mtb strains with a putative mechanism involving targeting of NDH-2 to prevent ATP synthesis of Mtb [16, 41, 42]. The phenothiazines were first examined by the medical community as a result of their anesthetic properties, and chlorpromazine was developed and widely used to treat neuropathy [43]. Subsequently, phenothiazines were repurposed for the treatment of TB [44, 45]. However, the safety issues of phenothiazines due to off-target effects led to their limited development as antituberculosis drugs [41]. Dunn et al. reported a type of alkyl-triphenyl-phosphonium (alkylTPP) compound capable of intracellular delivery to improve the localization and effective concentration of phenothiazine derivatives at the mycobacterial membrane. A representative compound (9)showed potent inhibition of Mtb with MIC = 0. 5 μg/mL [46].

|
Download:
|
| Fig. 3. Phenothiazine and phenazine analogues as NDH-2 inhibitors. | |
{kind=link}
The phenazine derivative clofazimine (10, Fig.3), an antileprosy drug, was also found to have potent antimycobacterial activity and was approved to treat TB [47]. Yano et al. demonstrated that clofazimine is reduced by NDH-2 as a redox cycling agent [48] via a reactive oxygen species (ROS) mechanism. Clofazimine enhances the activity of a second-line regimen against an isoniazid-resistant strain of Mtb in a mouse model [49]. It has been shown to act synergistically with bedaquiline and Q203 to block Mtb growth [50]. Unfortunately, clofazimine is clinically undesirable because it discolors the skin. To overcome this shortcoming, further SAR exploration led to discovery of a new generation compound TBI-166 (11), with efficacy and skin pigmentation potential better than that of clofazimine [51-53]. TBI-166 has been approved by the Chinese National Medical Products Administration (NMPA) to enter Phase I clinical trials.
In addition tophenothiazine and phenazines, AstraZeneca reported a series of quinolinyl pyrimidines as a promising class of NDH-2 inhibitors by HTS of an in-house library in 2012 (Fig.4) [54]. The representative compound (12) inhibits NDH-2 with IC50= 43 nmol/L, exhibits potent activity in vitro against Mtb with MIC = 0. 87 μg/mL, and has promising potential for further optimization. Another HTS campaign also disclosed two novel scaffolds, thioquinazoline and tetrahydroindazole, as selective inhibitors of NDH-2 [55]. CBR-1825 (13) and CBR-4032 (14) inhibit oxidative phosphorylation levels with IC50 values of 0. 14 μmol/L and 0. 5 μmol/L respectively, and inhibit the H37Ra strain with MIC50 values of 0. 43 and 6. 6 umol/L, respectively. Notably, these two compounds have no toxicity to mammalian cells. Further medicinal chemistry optimization of CRB-1825 (13) led to the most potent quinazoline derivative CBR-1922 (15) with MIC = 0. 09 umol/L against Mtb. Biochemical and genetic studies indicated that these two scaffoldshave differential inhibitory activities against the two homologous Ndh-2 enzymes Ndh and NdhA Mtb.

|
Download:
|
| Fig. 4. Quinolinyl-pyrimidine, thioquinazoline and tetrahydroindazole-based compounds as NDH-2 inhibitors. | |
{kind=link}
4.2. QcrB inhibitors 4.2.1. Imidazo[1,2-a]pyridine-3-carboxamides (IPAs)
The best characterized compounds targeting the cytochrome b subunit of the bcc complex (QcrB) are imidazopyridine amides (IPAs). Since Moraski et al. first discovered in 2011 that IPAs had potency and selectivity toward replication of the MDR and XDR Mtb strains [56], extensive research on this scaffold has been performed. In 2013, Pethe et al. reported an IPA-based compound, Q203 (19) as a promising antituberculosis drug candidate from phenotypic high-content screening (HCS) technology inside infected macrophages [25, 28]. The optimization was conducted by synthesis and evaluation of 477 derivatives [57]. Briefly, the discovery of Q203 started from a biphenyl compound (16), which was reported by Kang et al. (Fig.5) [58]. Further exploration of SAR at the imidazo[1,2-a]pyridine core and the para-position of the biphenyl compound led to compound 17, which is much more potent with extracellular MIC80= 0. 9 nmol/L and intracellular MIC80 = 0. 45 nmol/L and is metabolically stable, with human t1/2 = 83 min. However, subsequent in vivo pharmacokinetics (PK) studies suggested that 17 has strong lipophilicity, poor absorption, and rebounded in absorption after 4 h. In order to increase the solubility and reduce the lipophilicity, further optimization focused on the second benzene ring while retaining the 7-chloro-2-ethylimidazo[1,2-a]pyridine-3-carboxamide moiety. A 4-fluorophenyl-piperazine derivative (18) was found with MIC80 = 0. 3 nmol/L in antibacterial replication in macrophages, and good stability in mouse liver microsomes. Further optimization by replacing the fluorine with a trifluoromethoxy group led to a clinical candidate Q203 which displays unprecedented MIC80 values against Mtb H37Rv replicating both outside and inside the macrophage (extracellular MIC80 = 4. 0 nmol/L, intracellular MIC80 = 1.43 nmol/L) [27]. Q203 is efficacious in a mouse model of tuberculosis at a dose of < 1 mg/kg, which demonstrates its potency. Q203 has successfully entered Phase II clinical trials [59].

|
Download:
|
| Fig. 5. Discovery process of QcrB inhibitors Q203. | |
{kind=link}
Spontaneous-resistant mutants and site-directed amino acid mutation strategies demonstrated that Q203 targets the cyto-chrome bcc oxidoreductase b subunit (QcrB) by binding at the Qp site and thus interfering with ATP synthesis in the respiratory electron transport chain of Mtb. However, Q203 has only transient antimycrobacterial properties [60]. It was also shown that Q203 became bactericidal when the cytochrome bd oxidasegene was knocked out. Additionally, Matsoso et al. confirmed that the disabled assembly of cytochrome bcc or a genetic knock-out of cytochrome bcc in M. smegmatis failed to completely abolish bacterial growth [23]. Interestingly, Arora et al. found that when cytochrome bd oxidase was knocked out, Q203 restored the growth inhibition of Mtb [33]. Lu et al. confirmed that the presence of cytochrome bd inhibitors can enhance the effect of Q203 [61]. These results further indicated that the cytochrome bd oxidase also can maintain menaquinol oxidation in the presence of Q203 and a synthetic lethal pathway was observed by targeting both cytochrome bcc-aa3 and bd oxidases for antituberculosis drug development [60].
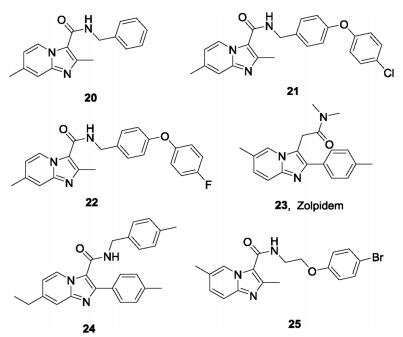
Other IPA derivatives studied as QcrB inhibitors are shown in Fig.6. Compound 20 was the first IPA compound with antitubercular activity reported by Moraski et al. in 2011 from screening of the compound library of Dow AgroScience. Compound 20 is active against Mtb with MIC90 values of 0.37–1.9 μmol/L in various assays, and is bacteriostatic against replicating Mtb [56]. However, there is no synergistic effect of compound 20 with other antituberculos drugs PA824, BTZ043, SQ109 [62]. Further medicinal chemistry optimization led to a series of imidazo[1,2-a]pyridines such as compounds 21 and 22, with dramatically enhanced potency and improved PK properties. The compounds have MIC values ≤ 0.006 μmol/L, and amongst them, 22 is most potent against the drug resistant strains with MICs < 0.03-0.8 μmol/L. Preliminary PK studies shows that it has ideal PK in mice with an AUC = 3850 ng h/mL and a T1/2 > 12 h [63]. In another study, the analogue 21 was selected as a promising lead compound with which to investigate its protective efficacy in a mouse model. Compared with untreated mice, the burden in the lungs and spleens in mice treated with 21 once daily 6 days per week for four weeks was significantly decreased [64]. Moraski et al. also reported zolpidem (23), a traditional medicine used to treat insomnia with a structure similar to that of IPA, exhibits moderate activity against Mtb with MIC = 10-50 mmol/L. The representative analogue (24) had outstanding in vitro potency against Mtb with MIC = 0.004 μmol/L [65]. Wu et al. reported a series of IPAs bearing an N-(2-phenoxyethyl) group with excellent activity in vitro against drug sensitive Mtb (MIC = 0.027-2 μg/mL) and two clinical MDR-TBs with MIC = 0.025-0.054 μg/mL [66]. The representative compound (25) showed acceptable safety and PK characteristics and served as a promising lead compound for further antituberculosis drug discovery.

|
Download:
|
| Fig. 6. Some reported IPAs studied as QcrB inhibitors. | |
{kind=link}
4.2.2. Pyrazolo [1,5-a] pyridines-3-carboxamides (PPA)
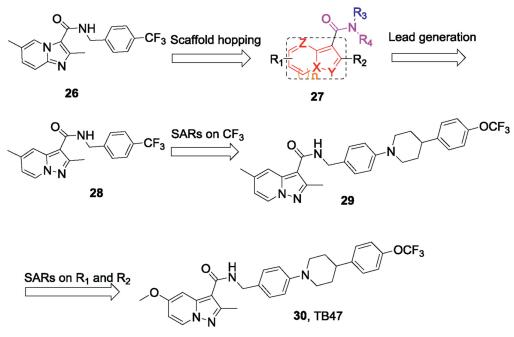
To pursue new antituberculosis drugs with alternative scaffolds, we designed a series of pyrazolo[1,5-a]pyridines-3-carbox-amides (PPAs) based on the IPA derivative (26) by using a scaffold hopping strategy (Fig.7) [67, 68]. A series of biheterocyclic variations on 27, including PPA, 1H-indole, benzofuran, pyrazolo [1,5-a]pyrimidine, pyrazolo[1,5-a]pyrimidin-5(4H)-one, imidazo [2,1-b]thiazole and pyrazolo[5,1-b]thiazole derivatives were synthesized and their antitubercular activities were evaluated. Encouragingly, we found the first designed compound (28) showed antimycobacterial activity against both H37Rv and H37Ra with MIC values of 69. 1 and 287. 9 nmol/L, respectively. Distancing of the para-CF3-bearing ring of 28 with large hydrophobic groups led to compound 29, with the identical lipophilic tail to that of Q203. Compound 29 showed strong antimycobacterial activity against H37Rv and H37Ra with MIC values of 7.7 and 5.7 nmol/L, respectively. Moreover, a VERO cell growth inhibition assay demonstrated 29 to be without cytotoxicity (IC50>100 μmol/L). Further investigation of the SARs of substituent derivatives of the PPA scaffolds afforded compound 30 (Fig.7), termed TB47 in a later study [69]. TB47 displayed excellent potency against H37Rv and H37Ra with MIC values of 11.1 and 5.6 nmol/L, respectively. TB47 is active against a panel of 56 Mtb clinical isolates (37 types of MDR-TB and 2 types of XDR-TB) with MIC values between 0.016 μg/mL and 0. 500 μg/mL. Pharmacokinetic and toxicity studies showed promising profiles, including negligible CYP450 interactions, cytotoxicity, and hERG channel inhibition [69]. Follow-up research into the in vivo antitubercular efficacy of TB47 was evaluated using a modified real-time monitoring noninvasive mouse model infected with a selectable marker-free auto-luminescent Mtb strain H37Ra. TB47 exhibited dose-dependent in vivo antitubercular activity and was well tolerated in all tested groups with no mortality. In particular, TB47 exhibited sustained bacteriostatic activity against Mtb H37Ra at a dose of 4 mg/kg/day [67]. The oxygen consumption assays indicated that TB47 inhibits oxygen consumption dependent on QcrB. Consistent with Q203, TB47 inhibits oxygen consumption only when the cytochrome bd was deleted. However, TB47 is synergistic with the clinically available drugs pyrazinamide and rifampicin in vivo models, suggesting a promising role in combination therapies [69]. TB47 is currently in preclinical studies [59].

|
Download:
|
| Fig. 7. Optimization process of TB47. | |
{kind=link}
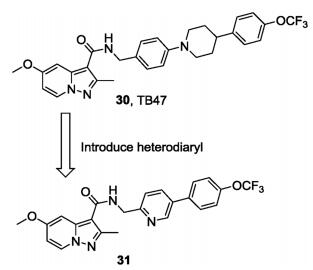
Subsequently, another series of PPA derivatives bearing a diphenyl or a heterodiaryl side chain was designed with an optimization strategy similar to that in which TBA354 was based on TB47, aiming to improve the efficacy toward drug resistant Mtb and search for a back-up candidate (Fig.8) [70, 71]. One of the most potent compounds (31) exhibits excellent in vitro potency (MIC < 0. 002 μg/mL) against the drug susceptible H37Rv strain and drug resistant Mtb strains (INH-resistant (rINH), MIC < 0. 002 μg/mL; RMP-resistant (rRMP), MIC = 0. 002 μg/mL). Compound 31 also displayed good PK profiles with oral bioavailability of 41% and significantly reduced the bacterial burden in an autoluminescent H37Ra infected mouse model, suggesting it could be a new lead for further discovery of antitubercular drugs [71].

|
Download:
|
| Fig. 8. Optimization strategy of compound 31 based on TB47. | |
{kind=link}
4.2.3. Lansoprazole (LPZ)
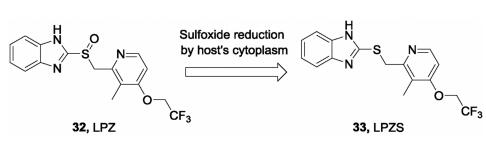
In 2015, Rybniker et al. disclosed that a gastric protonpump inhibitor, lansoprazole (LPZ, 32) as a prodrug, possessed intracellular antimycobacterial activity (Fig.9) [72]. However, other proton pump inhibitors such as omeprazole and pantoprazole do not possess this effect. By reduction of its sulfoxide, LPZ is converted into its active form, lansoprazole sulfide (LPZS, 33) in the cytoplasm of the host and then exhibits antitubercular activity. LPZS failed to change into the sulfenic acid form, which suggests its selectivity over the human H+K+-ATPase. The whole-genomese-quencing and site-directed mutation assay revealed that LPZS achieves the antitubercular effect by targeting QcrB of Mtb. Moreover, LPZS is highly active against drug-resistant clinical strains due to its unique mechanism of action. This study provides a valuable proof of concept of host-mediated prodrug activation for antibacterial drug discovery.

|
Download:
|
| Fig. 9. Sulfoxide reduction by host cells converts LPZ to LPZS. | |
{kind=link}
4.2.4. Other types
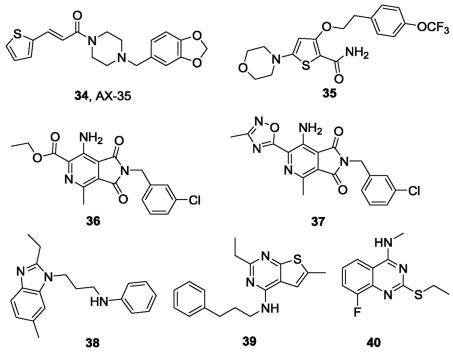
In addition to the aforementioned IPAs, PPAs and LPZ, other reported QcrB inhibitors are shown in Fig.10. One thiophenevi-nylpiperazine amide AX-35 (34), identified through phenotypic screening conducted by GlaxoSmithKline (GSK) [73], has potent activity against Mtb H37Rv with a MIC value of 0. 05 μg/mL [74]. The multifaceted validation studies, including allele exchange, transcriptome profiling and bioflux assays, demonstrated that AX-35 targets QcrB with a binding mode different from that of Q203. Similar to Q203, AX-35 shows bactericidal activity in the absence of cytochrome bd oxidase. Nevertheless, the study provides a promising scaffold for antituberculosis drug development. Another novel scaffold, morpholino thiophene that targets QcrB has been reported [75]. The representative compound (35) has considerable activity both in vitro against Mtb H37Rv (MIC = 0. 2 μmol/L) and in vivo in an acute murine infection model of Mtb. Interestingly, HTS by Novartis identified a pyrrolo[3,4-c]pyridine-1,3-dione (36) with good in vitro activity against Mtb (MIC = 0. 132 μmol/L) [76]. Further SAR optimization by replacing the ester with a methyl oxadiazole led to the most potent compound (37) with MIC = 65 nmol/L. Mode of action studies demonstrated that this series of compounds targeted QcrB and was hypersusceptible to cytochrome bd deletion mutant. More recently, the novel scaffolds alkylbenzimidazole (38) [77], 4-amino-thieno[2,3-d] pyrimidine (39) [78] and 2-ethylthio-4-methylaminoquinazoline (40) [79] with nanomolar activities were also reported as potential antituberculosis drugs targeting QcrB. The 2-ethylthio-4-methylaminoquinazoline scaffold binds differently from that of Q203 by targeting two subunits of cytochrome bcc(QcrA and QcrB) Mtb.

|
Download:
|
| Fig. 10. Other reported QcrB inhibitors. | |
{kind=link}
4.3. ATP synthase inhibitors 4.3.1. Bedaquiline(TMC207)
The diarylquinoline compound, bedaquiline (1) discovered by whole-cell screening of prototypes, is the first new antitubercular drug approved for the treatment of MDR-TB in nearly 40 years [4]. Bedaquiline exhibits potent activity against drug-sensitive and drug-resistant Mtb with MIC value of 0. 06 μg/mL, and has high activity in both replicating and dormant mycobacteria. Remark-ably, bedaquiline, alone or combination, is more effective than isoniazid and rifampin in vivo. The X-ray crystal structure and functional assays demonstrated that bedaquiline specifically binds in the ion-binding sites of the Fo rotor ring (the c-ring) in ATP synthase to block the ion translocation and ATP synthase function (Fig.11A) [40, 80]. The studies of the c-rings of ATP synthase are of the M. phleistrain, which shares 83. 7% identity with that of Mtb. Bedaquiline covers a large surface of the c-rings (~135 Å) by forming extensive van der Waals interactions with nine residues Gly62, Leu63, Glu65, Ala66, Ala67, Tyr68, Phe69, Ile70 and Leu72, mainly located in the two adjacent c-rings (Fig.11A). The hydroxyl group makes an indirect hydrogen bond with Glu65 mediated by one water, and the protonated dimethylamino group extends into the ion-binding site where it forms an intermolecular hydrogen bond with Glu65 (Fig.11B). These observed interactions explain the strong bactericidal activity of bedaquiline against Mtb.

|
Download:
|
| Fig. 11. The X-ray structure of the c-rings of ATP synthase from M. phlei bound to bedaquiline. (A) The cartoon represents the structure of the c-rings complexed with bedaquiline (PDB ID: 4V1F). Details of the binding interactions between bedaquiline and c-rings of ATP synthase are shown in the right insert. The key residues are represented as sticks and labels. Hydrogen bonds are shown as yellow dashes. (B) Two dimensional (2D) plot of bedaquiline with key residues of c-rings. The purple arrow, blue straight line and gray shaded portion indicate hydrogen bonds, ion-ion and van der Waals interactions, respectively. | |
{kind=link}
Bedaquiline shows a cardiotoxic side effect due to prolonged QT intervals, between Q and T waves in the heart, caused by inhibition of cardiac human ether-a-go-related gene (hERG) potassium ion channels (IC50 = 0. 2 μg/mL) [81], limiting its wide application labeled and leading to a black-box warning in clinical practice [5]. Additionally, the high lipophilicity of bedaquiline (clogP = 7. 25)also led to the long half-life (5–6 months) and phospholipidosis side effects [82]. Thus, bedaquiline analogues with similar antimycobacterial activity and lower clogP and less inhibition of the hERG would be of particular interest.
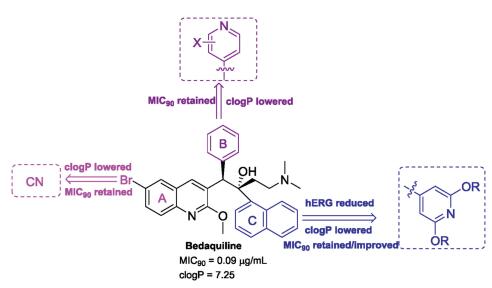
4.3.2. Bedaquiline analoguesTo the best of our knowledge, many bedaquiline analogues with an improved safety profile have been reported by medicinal chemists since the discovery of bedaquiline [83]. Combined with the structure features of bedaquiline which are responsible for the potent activity and toxicity, the modifiable positions of bedaquiline are mainly focused on the substituent at the 6-position of the quinoline A-ring, the phenyl B-ring and the naphthalene C ring (Fig.12). The dimethylamine moiety largely contributes to hERG inhibition, while replacing it with less basic group (pKa < 8) significantly decreased the potency [84]. Shortening or lengthening the dimethylethylamine chain led to more cytotoxicity due to the increased lipophilicity. Thus, the dimethylethylamine moiety was left unchanged in subsequent optimizations.

|
Download:
|
| Fig. 12. The optimization positions of bedaquiline and the corresponding preferred groups. | |
{kind=link}
The scientists at University of Auckland have done much research in this area. In 2017, Tong et al. investigated the SARs of substituents at the 6-position of the A-ring and first disclosed that a cyano group provided a positive correlation between the lipophilicity and potency [85]. The representative compounds (41 and 42) significantly decreased the clogP while displayed minimal effects on MIC90 (Fig.13). In the same year, the same research group reported another series of bedaquiline analogues in which the phenyl B-ring was replaced with monocyclic heterocycles such as thiophene, furan and pyridine [86]. Among them, only the 4-pyridyl analogues displayed comparable antimycobac-terial activity. Compound 43 was most active against Mtb in both MABA and LORA assays with MICs < 0. 02 μmol/L and its clogP value was significantly lower than that of bedaquiline (Fig.13). Sutherland et al. synthesized and evaluated the c-ring substituted BDQ derivatives, which replaced the lipophilic naphthalene with various bicyclic heterocycles and a substituted pyridinel ring [87, 88]. The representative 7-benzofuran analogue (44) retains the potency and has improved hERG inhibition while still displaying high clogP. Most of substituted pyridyl analogues show a lower clogP of about of 5 and retain the antitubercular activity [88]. However, they still show potent inhibition against the hERG potassium channel comparable to that of bedaquiline. Only recently, it was found that the analogues in which the naphthalene was replaced by a 3,5-dialkoxy-4-pyridyl group retain high antitubercular activity albeit with a lower clogP and less inhibition against hERG than bedaquiline [89, 90]. Two of these analogues 45 (TBAJ-876) and 46 (TBAJ-587) exhibit ideal in vitro and in vivo potency and have entered preclinical evaluation (Fig.13). Further biochemical and NMR titration studies demonstrated that TBAJ-876 retains bedaquiline's MOA bonding to the c and ε subunits of ATP synthase of Mtb [91]. These studies indeed provide hope to overcome the challenges in tuberculosis treatment.
|
|
Download:
|
| Fig. 13. Representative bedaquiline analogues. | |
4.3.3. Other types
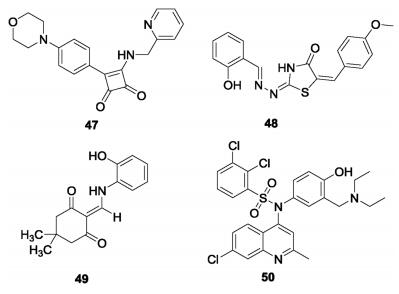
In addition to bedaquiline, some other potential scaffolds have been reported to target ATP synthase of Mtb (Fig.14). Squaramide (47), discovered by AstraZeneca using HTS, is a nanomolar potent (IC50 = 30 nmol/L) inhibitor against ATP synthase and is active against Mtb with MIC = 0. 5 μmol/L [92]. Analysis of spontaneous resistant mutants and molecular docking studies indicated a different binding site for squaramide on ATP synthase compared to bedaquiline, which was proposed by its interface with subunits a and c of ATP synthase. Further, the demonstrated in vivo antitubercular efficacy showed it to be a promising lead for further development. Kumar et al. screened 700 compounds and found two compounds, 5228485 (48) and 5220632 (49) with IC50 values of 0. 32 and 4. 0 μg/mL respectively against ATP synthase of M. smegmatis [93]. Both 48 and 49 showed excellent antitubercular activity with MICs of 0. 5–2. 0 μg/mL against Mtb H37Rv. Modeling studies suggested that their binding mode with ATP synthase involves especiallythe phenol group of compound 48, which forms a strong hydrogen bond with Leu59. This provides ideas for the subsequent structural design. Another quinoline sulfonamide (50) was designed by hit to lead optimization and evaluated as a selective potent mycobacterium ATPase inhibitor with IC50 = 0. 51 μmol/L [94]. Further pharmacokinetic and in vivo efficacy studies suggested it is a promising candidate for further study.

|
Download:
|
| Fig. 14. Other reported scaffolds as inhibitors of ATP synthase. | |
{kind=link}
5. Conclusions
A critical unmet clinical need to combat TB epidemics is the development of potenttherapeutic agents with new mechanisms of action to reduce the treatment time of MDR-TB and XDR-TB. With the approval of the ATP synthase inhibitor bedaquiline and the discovery of the candidate Q203 that targets QcrB, it has been shown that interference with the oxidative phosphorylation pathway for energy generation is a promising antituberculosis strategy. As mentioned above, many new scaffold inhibitors targeting the key enzymes (NDH-2, QcrB and ATP synthase) in the respiratory chain of Mtb have been developed and evaluated as potential antituberculosis drugs. In spite of the success of targeting the electron transport chain pathway to prevent the synthesis of ATP in Mtb, some challenges remain to be addressed.
First, compounds targeting QcrB can significantly inhibit the growth of Mtb at lower nanomolar concentrations, but theylack sustained bacteriostatic activity. The high expression of cytochrome bd oxidase in Mtb can partially reduce the efficacy of cytochrome bcc inhibitors. With reports that cytochrome bd inhibitors can enhance the antimycobacterial activity of the cytochrome bcc inhibitor Q203, it is indicated that a drug combination targeting cytochrome bcc and cytochrome bd may represent the cornerstone of a complementary sterilizing drug combination for the treatment of TB. Second, although the approved drug bedaquiline shows good therapeutic effect against MDR-TB, it is cardiotoxic and phospholipidosis side effects are a serious problem. Thus, it is urgent to continue efforts to develop new agents that selectively target the respiratory chain to avoid any toxicity or other unexpected effects to host cells. Third, at present, only the inhibitors of these three enzymes NDH-2, QcrB and ATP synthase in the respiratory chain have been well characterized. These facts highlight the need for further exploration of underexplored components like SDH and cytochrome bd oxidase to discover more potent inhibitors as alternate antituberculosis agents. Finally, there is no positive correlation between the antimycobacterial activity and physicochemical properties of most compounds reported to target the respiratory chain, due to the fact that they are obtained from the whole-cell based HTS but lack target-based rational drug design. The disclosure of the X-ray crystal structure of F1Fo-ATP synthase bound with bedaquiline and cryo-EM structure of the cytochrome bcc-aa3 supercomplex as well as E. coli cytochrome bd oxidase will greatly aid the future investigations of rationally designing next generation inhibitors. Nevertheless, the continued drug discovery efforts targeting the oxidative phosphorylation pathway in respiratory chain may provide novel strategies with which to treat MDR-TB and XDR-TB.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors appreciate the financial support from the National Natural Science Foundation of China (No. 81922062), Guangdong Provincial Science and Technology Program (No. 2018A050506043) and Jinan University.
| [1] |
T. Wirth, F. Hildebrand, C. Allix-Beguec, et al., PLoS Pathog 4 (2008) e1000160. DOI:10.1371/journal.ppat.1000160 |
| [2] |
World Health Organization, Global Tuberculosis Report 2019 (WHO), https://www.who.int/tb/publications/global_report/en/.
|
| [3] |
L. Pitance, L. Vecellio, T. Leal, et al., J. Aerosol. Med. Pulm. D. 23 (2010) 389-396. DOI:10.1089/jamp.2010.0816 |
| [4] |
K. Andries, P. Verhasselt, J. Guillemont, et al., Science 307 (2005) 223-227. DOI:10.1126/science.1106753 |
| [5] |
A. H. Diacon, P. R. Donald, A. Pym, et al., Antimicrob Agents Ch. 56 (2012) 3271-3276. DOI:10.1128/AAC.06126-11 |
| [6] |
E. Cox, K. Laessig, N. Engl. J. Med. 371 (2014) 689-691. DOI:10.1056/NEJMp1314385 |
| [7] |
M. T. Gler, V. Skripconoka, E. Sanchez-Garavito, et al., N. Engl. J. Med. 366 (2012) 2151-2160. DOI:10.1056/NEJMoa1112433 |
| [8] |
C. D. Tweed, R. Dawson, D. A. Burger, et al., Lancet Respir. Med. 7 (2019) 1048-1058. DOI:10.1016/S2213-2600(19)30366-2 |
| [9] |
C. G. Mohan, Impact of Target-Based Drug Design in Anti-bacterial Drug Discovery for the Treatment of Tuberculosis, Springer (2019) 307-346. |
| [10] |
K. Mdluli, T. Kaneko, A. Upton, Cold Spring Harb. Perspect. Med. 5 (2015) a021154. DOI:10.1101/cshperspect.a021154 |
| [11] |
S. L. Tran, G. M. Cook, J. Bacteriol. 187 (2005) 5023-5028. DOI:10.1128/JB.187.14.5023-5028.2005 |
| [12] |
C. A. Kerantzas, W. R. Jacobs Jr. , mBio 8 (2017) e01586-16. |
| [13] |
G. Sotgiu, G. Sulis, A. Matteelli, Microbiol. Spectr. 5 (2017). |
| [14] |
D. Bald, A. Koul, FEMS Microbiol. Lett. 308 (2010) 1-7. DOI:10.1111/j.1574-6968.2010.01959.x |
| [15] |
S. Kerscher, S. Dröse, V. Zickermann, U. Brandt, ResultsProbl. CellDiffer. 45 (2008) 185-222. |
| [16] |
E. A. Weinstein, T. Yano, L. S. Li, et al., Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 4548-4553. DOI:10.1073/pnas.0500469102 |
| [17] |
G. Unden, J. Bongaerts, Biochim. Biophys. Acta 1320 (1997) 217-234. DOI:10.1016/S0005-2728(97)00034-0 |
| [18] |
S. P. S. Rao, S. Alonso, L. Rand, T. Dick, K. Pethe, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 11945-11950. DOI:10.1073/pnas.0711697105 |
| [19] |
L. Miesel, T. R. Weisbrod, J. A. Marcinkeviciene, R. Bittman, W. R. Jacobs Jr. , J. Bacteriol. 180 (1998) 2459-2467. DOI:10.1128/JB.180.9.2459-2467.1998 |
| [20] |
D. Bald, C. Villellas, P. Lu, A. Koul, mBio 8 (2017) e00272-17. |
| [21] |
C. Pidathala, R. Amewu, B. Pacorel, et al., J. Med. Chem. 55 (2012) 1831-1843. |
| [22] |
R. Cox, G. Cook, Curr. Mol. Med. 7 (2007) 231-245. DOI:10.2174/156652407780598584 |
| [23] |
L. G. Matsoso, B. D. Kana, P. K. Crellin, et al., J. Bacteriol. 187 (2005) 6300-6308. DOI:10.1128/JB.187.18.6300-6308.2005 |
| [24] |
H. Gong, J. Li, A. Xu, et al., Science 362 (2018) eaat8923. DOI:10.1126/science.aat8923 |
| [25] |
K. A. Abrahams, J. A. Cox, V. L. Spivey, et al., PLoS One 7 (2012) e52951. DOI:10.1371/journal.pone.0052951 |
| [26] |
P. A. Mak, S. P. Rao, M. P. Tan, et al., ACS Chem. Biol. 7 (2012) 1190-1197. DOI:10.1021/cb2004884 |
| [27] |
S. Kang, R. Y. Kim, M. J. Seo, et al., J. Med. Chem. 57 (2014) 5293-5305. |
| [28] |
K. Pethe, P. Bifani, J. Jang, et al., Nat. Med. 19 (2014) 1157-1160. |
| [29] |
V. B. Borisov, R. Murali, M. L. Verkhovskaya, et al., Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 17320. DOI:10.1073/pnas.1108217108 |
| [30] |
V. B. Borisov, R. B. Gennis, J. Hemp, M. I. Verkhovsky, Biochim. Biophys. Acta 1807 (2011) 1398-1413. DOI:10.1016/j.bbabio.2011.06.016 |
| [31] |
L. Shi, C. D. Sohaskey, B. D. Kana, et al., Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 15629. DOI:10.1073/pnas.0507850102 |
| [32] |
P. Lu, M. Heineke, A. Koul, et al., Sci. Rep. 5 (2015) 10333. DOI:10.1038/srep10333 |
| [33] |
K. Arora, B. Ochoa-Montano, P. S. Tsang, et al., Antimicrob. Agents Chemother. 58 (2014) 6962-6965. DOI:10.1128/AAC.03486-14 |
| [34] |
C. von Ballmoos, G. Cook, P. Dimroth, Ann. Rev. Biophys. 37 (2008) 43-64. DOI:10.1146/annurev.biophys.37.032807.130018 |
| [35] |
D. Pogoryelov, O. Yildiz, J. D. Faraldo-Gomez, T. Meier, Nat. Struct. Mol. Biol. 16 (2009) 1068-1073. DOI:10.1038/nsmb.1678 |
| [36] |
M. Diez, B. Zimmermann, M. Börsch, et al., Nat. Struct. Mol. Biol. 11 (2004) 135-141. DOI:10.1038/nsmb718 |
| [37] |
H. Noji, R. Yasuda, M. Yoshida, K. Kinosita Jr. , Nature 386 (1997) 299-302. DOI:10.1038/386299a0 |
| [38] |
J. K. Hakulinen, A. L. Klyszejko, J. Hoffmann, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) e2050-2056. DOI:10.1073/pnas.1203971109 |
| [39] |
A. C. Haagsma, I. Podasca, A. Koul, et al., PLoS One 6 (2011) e23575. DOI:10.1371/journal.pone.0023575 |
| [40] |
A. Koul, N. Dendouga, K. Vergauwen, et al., Nat. Chem. Biol. 3 (2007) 323-324. DOI:10.1038/nchembio884 |
| [41] |
D. Ordway, M. Viveiros, C. Leandro, et al., L. Antimicrob Agents Chemother. 47 (2003) 917-922. DOI:10.1128/AAC.47.3.917-922.2003 |
| [42] |
S. Sellamuthu, M. Singh, A. Kumar, S. K. Singh, Expert. Opin. Ther. Tar. 21 (2017) 559-570. DOI:10.1080/14728222.2017.1327577 |
| [43] |
M. Kaminska, Folia med Craco. 9 (1967) 115-143. |
| [44] |
T. Yano, L. S. Li, E. Weinstein, J. S. Teh, H. Rubin, J. Biol. Chem. 281 (2006) 11456-11463. DOI:10.1074/jbc.M508844200 |
| [45] |
M. Viveiros, S. Bosne-David, L. Amaral, Int. J. Antimicrob. Agents 16 (2000) 69-71. DOI:10.1016/S0924-8579(00)00199-0 |
| [46] |
E. A. Dunn, M. Roxburgh, L. Larsen, et al., Bioorg. Med. Chem. 22 (2014) 5320-5328. DOI:10.1016/j.bmc.2014.07.050 |
| [47] |
B. Lechartier, S. T. Cole, AntimicrobAgentsChemother 59 (2015) 4457-4463. |
| [48] |
T. Yano, S. Kassovska-Bratinova, J. Teh, et al., J. Biol. Chem. 286 (2011) 10276-10287. DOI:10.1074/jbc.M110.200501 |
| [49] |
J. H. Grosset, S. Tyagi, D. V. Almeida, et al., Am. J. Respir. Crit. Care Med. 188 (2013) 608-612. DOI:10.1164/rccm.201304-0753OC |
| [50] |
D. A. Lamprecht, P. M. Finin, M. A. Rahman, et al., Nat. Commun. 7 (2016) 12393-12393. DOI:10.1038/ncomms12393 |
| [51] |
D. Zhang, Y. Lu, K. Liu, et al., J. Med. Chem. 55 (2012) 8409-8417. |
| [52] |
D. Zhang, Y. Liu, C. Zhang, et al., Molecules 19 (2014) 4380-4394. DOI:10.3390/molecules19044380 |
| [53] |
J. Xu, B. Wang, L. Fu, et al., Antimicrob. Agents Chemother. 63 (2019) e02155-02118. |
| [54] |
P. Shirude, B. Paul, N. Choudhury, et al., ACS Med. Chem. Lett. 3 (2012) 736-740. DOI:10.1021/ml300134b |
| [55] |
M. B. Harbut, B. Yang, R. Liu, et al., Angew. Chem. Int. Ed. Engl. 57 (2018) 3478-3482. DOI:10.1002/anie.201800260 |
| [56] |
G. C. Moraski, L. D. Markley, P. A. Hipskind, et al., ACS Med. Chem. Lett. 2 (2011) 466-470. DOI:10.1021/ml200036r |
| [57] |
Z. No, J. Kim, P. B. Brodin, et al., WO2011113606A1 (2011). |
| [58] |
S. Kang, R. Y. Kim, M. J. Seo, et al., J. Med. Chem. 57 (2014) 5293-5305. |
| [59] | |
| [60] |
N. P. Kalia, E. J. Hasenoehrl, N. B. Ab Rahman, et al., Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 7426-7431. DOI:10.1073/pnas.1706139114 |
| [61] |
P. Lu, A. Asseri, M. Kremer, et al., Sci. Rep. 8 (2018) 2625. DOI:10.1038/s41598-018-20989-8 |
| [62] |
T. O'Malley, T. Alling, J. V. Early, et al., Antimicrob. Agents Chemother. 62 (2018) e02439-17. |
| [63] |
G. C. Moraski, L. D. Markley, J. Cramer, et al., ACS Med. Chem. Lett. 4 (2013) 675-679. DOI:10.1021/ml400088y |
| [64] |
Y. Cheng, G. C. Moraski, J. Cramer, M. J. Miller, J. S. Schorey, PLoS One 9 (2014) e87483. DOI:10.1371/journal.pone.0087483 |
| [65] |
G. C. Moraski, P. A. Miller, M. A. Bailey, et al., ACS Infect. Dis. 1 (2015) 85-90. |
| [66] |
Z. Wu, Y. Lu, L. Li, et al., ACS Med. Chem. Lett. 7 (2016) 1130-1133. DOI:10.1021/acsmedchemlett.6b00330 |
| [67] |
J. Tang, B. Wang, T. Wu, et al., ACS Med. Chem. Lett. 6 (2015) 814-818. DOI:10.1021/acsmedchemlett.5b00176 |
| [68] |
X. Lu, J. Tang, Z. Liu, et al., Bioorg. Med. Chem. 26 (2016) 5916-5919. DOI:10.1016/j.bmcl.2016.11.003 |
| [69] |
X. Lu, Z. Williams, K. Hards, et al., ACS Infect. Dis. 5 (2019) 239-249. DOI:10.1021/acsinfecdis.8b00225 |
| [70] |
A. M. Upton, S. Cho, T. J. Yang, et al., Antimicrob. Agents Chemother. 59 (2015) 136-144. DOI:10.1128/AAC.03823-14 |
| [71] |
X. Hu, B. Wan, Y. Liu, et al., ACS Med. Chem. Lett. 10 (2015) 295-299. |
| [72] |
J. Rybniker, A. Vocat, C. Sala, et al., Nat. Commun. 6 (2015) 7659. DOI:10.1038/ncomms8659 |
| [73] |
L. Ballell, R. H. Bates, R. J. Young, et al., ChemMedChem 8 (2013) 313-321. DOI:10.1002/cmdc.201200428 |
| [74] |
C. S. Foo, A. Lupien, M. Kienle, et al., mBio. 9 (2018) e01276-18. |
| [75] |
L. A. T. Cleghorn, P. C. Ray, J. Odingo, et al., J. Med. Chem. 61 (2018) 6592-6608. |
| [76] |
R. van der Westhuyzen, S. Winks, C. R. Wilson, et al., J. Med. Chem. 58 (2015) 9371-9381. |
| [77] |
N. S. Chandrasekera, T. Alling, M. A. Bailey, et al., J. Med. Chem. 58 (2015) 7273-7285. |
| [78] |
A. E. M. B. Gregory, A. Harrison, M. Singh, et al., mSphere 4 (2019) e00606-19. |
| [79] |
A. Lupien, C. S. Y. Foo, S. Savina, et al., PLoS Pathog. 16 (2020) e1008270. DOI:10.1371/journal.ppat.1008270 |
| [80] |
L. Preiss, J. D. Langer, Ö. Yildiz, et al., Sci. Adv. 1 (2015) e1500106. DOI:10.1126/sciadv.1500106 |
| [81] |
E. B. Chahine, L. R. Karaoui, H. Mansour, Ann. Pharmacother. 48 (2014) 107-115. DOI:10.1177/1060028013504087 |
| [82] |
A. K. Kakkar, N. Dahiya, Tuberculosis (Edinb) 94 (2014) 357-362. DOI:10.1016/j.tube.2014.04.001 |
| [83] |
H. Patel, R. Pawara, K. Pawara, et al., Tuberculosis (Edinb) 117 (2019) 79-84. DOI:10.1016/j.tube.2019.06.005 |
| [84] |
J. Guillemont, C. Meyer, A. Poncelet, X. Bourdrez, K. Andries, Future Med. Chem. 3 (2011) 1345-1360. DOI:10.4155/fmc.11.79 |
| [85] |
A. S. T. Tong, P. J. Choi, A. Blaser, et al., ACS Med. Chem. Lett. 8 (2011) 1019-1024. |
| [86] |
P. J. Choi, H. S. Sutherland, A. S. T. Tong, et al., Bioorg. Med. Chem. 27 (2017) 5190-5196. DOI:10.1016/j.bmcl.2017.10.042 |
| [87] |
H. S. Sutherland, A. S. T. Tong, P. J. Choi, et al., Bioorg. Med. Chem. 26 (2018) 1797-1809. DOI:10.1016/j.bmc.2018.02.026 |
| [88] |
A. Blaser, H. S. Sutherland, A. S. T. Tong, et al., Bioorg. Med. Chem. 27 (2019) 1283-1291. DOI:10.1016/j.bmc.2019.02.025 |
| [89] |
H. S. Sutherland, A. S. T. Tong, P. J. Choi, et al., Bioorg. Med. Chem. 27 (2019) 1292-1307. DOI:10.1016/j.bmc.2019.02.026 |
| [90] |
H. S. Sutherland, A. S. T. Tong, P. J. Choi, et al., Bioorg. Med. Chem. 28 (2020) 115213. DOI:10.1016/j.bmc.2019.115213 |
| [91] |
J. P. Sarathy, P. Ragunathan, J. Shin, et al., Antimicrob. Agents Chemother. 63 (2019) e01191-01119. |
| [92] |
S. J. Tantry, S. D. Markad, V. Shinde, et al., J. Med. Chem. 60 (2017) 1379-1399. |
| [93] |
S. Kumar, R. Mehra, S. Sharma, et al., Tuberculosis (Edinb). 108 (2018) 56-63. DOI:10.1016/j.tube.2017.10.008 |
| [94] |
S. Singh, K. K. Roy, S. R. Khan, et al., Bioorg. Med. Chem. 23 (2015) 742-752. DOI:10.1016/j.bmc.2014.12.060 |