 2020, Vol. 31
2020, Vol. 31 


b Zhejiang Zhong Cheng Medicine Co., Ltd., Hangzhou 310012, China
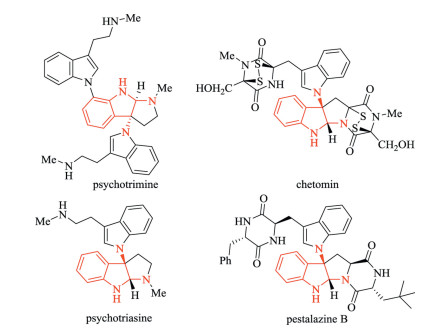
2, 3-Diamino indoline, an important vicinal diamine, is a very common moiety in natural products and pharmaceutics, especially in alkaloids. The methodology for its preparation, which would enable the study of its biological functions and the development of related alkaloid drugs, has attracted significant interest (Fig. 1) [1-4]. For example, the alkaloid psychotrimine has been isolated from Psychotria rostrat by Takayama and exhibits high antifungal activity via membrane disruption. Thus, green and efficient methods for building 2, 3-diaminoindoline groups are highly desirable.

|
Download:
|
| Fig. 1. Natural products containing 2, 3-diaminoindoline. | |
During the past decade, difunctionalization of C=C double bonds has emerged as a valuable and efficient synthetic approach [5-8]. In comparison to direct diamination, the diazido-process has attracted more interest. This is due to the easy conversion of diazido products to vicinal diamines. In addition, the dearomatization of indoles via nitrogen transfer involves a radical species, and azide radicals are easier to form than amine radicals [9, 10]. Vicinal diazides are useful synthons for heterocylic biologically active compounds [11], and play an important role in drug discovery and chemical biology due to their use in click chemistry [12-14]. Thus far, several methodologies have been successfully developed for the construction of 2, 3-diazidoindoline. Yasumitsu et al. developed an efficient method for the diazidation of indoles using azidoiodinane with a 7:2 ratio of diastereomers [15].
The group of Moriarty has reported the diazidation of indole using iodosobenzene with a 34% yield (Scheme 1a) [16]. Xu and coworkers have developed a diastereoselective (d.r. up to 20:1) diazidation of indole catalyzed by an iron complex with chiral ligands [17]. Vincent and co-workers have reported the synthesis of 2, 3-diazido indoline via electrochemistry in 41%-79% yield (Scheme 1b) [18]. However, these methods have some limitations, including low yields, poor diastereoselectivities, harsh conditions, the requirement for organic solvents, expensive ligands and complex reactor setups. Thus, conducting the highly stereoselective, efficient 2, 3-diazidation of indole under mild conditions is still a significant challenge.

|
Download:
|
| Scheme 1. Diazidation of double bonds in indoles. | |
Herein, we report a directing-group-assisted, stereoselective 2, 3-diazidation of indole via the copper-catalyzed direct dearomatization of indoles.
This method can be used for a broad range of indoles, with good yields and excellent diastereoselectivities in aqueous solution (Scheme 1). Additionally, this novel method enables the late-stage modification of indole-containing bioactive molecules such as tryptophan and 3-indoleacetic acid.
In recent years, directing groups have been shown to greatly improve the reactivity and stereoselectivity of C-H activation [19-26]. Various directing groups have been developed for the dearomatization of indoles, giving indolin-3-ones and cyclopropanated indolines in high yields [27-30]. Inspired by these results, we hypothesized that directing groups could also promote the formation of 2, 3-diazido indoline via the diazidation of indoles. Thus, we commenced our study by using 2-pyrimidyl as the directing group, and initially screened transition metal catalysts. Among the tested catalysts, Cu(OAc)2 gave the desired product in 30% yield (Table 1, entries 1-6). Then PhIO, PIFA, DCP, H2O2, K2S2O8, TBHP and MnO2 were considered as oxidants. The results showed that [PhI(OAc)2] was the most effective oxidant (Table 1, entries 7- 13).
|
|
Table 1 Optimization of the reaction conditions.a |

{kind=link}
{kind=link}
To further improve the product yield, different azide sources were screened, and the use of TMSN3 greatly increased the yield to 60% (Table 1, entry 14). Subsequently, the effect of the solvent on the reaction was studied using organic solvents including DMSO, dioxane, THF, DCE, DCM, MeOH, and MeCN. None of these solvents improved the yields with the exception of acetone (Table S1 in Supporting information), for which the yield increased to 85% (Table 1, entry 15). To explore the environmental friendliness of the methodology, the reaction was carried out in aqueous media. Similarly to the literatures, the desired product 3 was successfully obtained [31-33]. Interestingly, the yield increased slightly to 90% and reaction time decreased to 40 min (water:acetone = 4:1) (Table 1, entry 16). However, 3 was formed in 30% yield and more than 60% of 1a was recovered when using water as single solvent (Table 1, entry 17). Thus, 0.3 mmol of 1, 0.9 mmol of 2, 0.9 mmol of [PhI(OAc)2], in aqueous solution 2.5 mL (contained 20% acetone) at 25 ℃ for 40 min were chosen as the optimal reaction conditions for the directed diazidation.
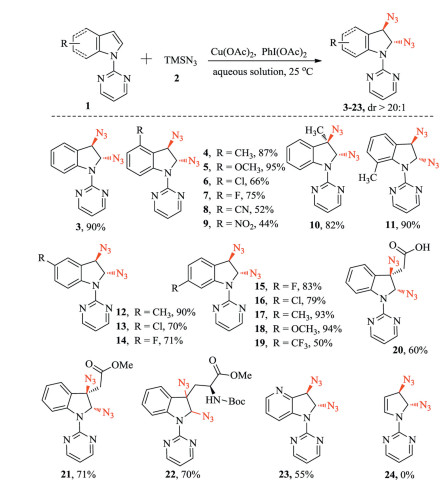
With the optimized reaction conditions in hand, a range of different substituted indoles with 2-pyrimidyl groups were investigated. As shown in Scheme 2, all of the functional groups in indole at different positions (C3- to C7-) were well tolerated in the reaction, and the desired products (3-19) were produced in moderate to excellent yields (44%-90%). Electron-donating groups, such as methyl and methoxy (4, 5, 10, 11, 12, 17, 18) offered higher yields than electron-withdrawing groups (6-9, 13-16, 19).

|
Download:
|
| Scheme 2. Scope of indoles in the 2, 3-diazidation. Reaction conditions: 1a (0.3 mmol), (0.9 mmol), Cu(OAc)2·H2O (0.06 mmol), PhI(OAc)2 (0.9 mmol), aqueous solution 2.5 mL (contained 20% acetone) at 25 ℃ for 40 min. Isolated yields. | |
{kind=link}
Then, we studied the applicability of the methodology to natural compounds. After the installation of the pyrimidyl group as a directing group, indole-3-acetic acid, a ubiquitous endogenous growth hormone in plants, can undergo the reaction to obtain the desired product (20) and its derivative (21) in 60% and 71% yield, respectively. The natural amino acid tryptophan can also tolerate this reaction and gave 22 in 70% yield. Furthermore, 4-azaindole with an added pyrimidyl group also afforded the expected product 23 in 55% yield, while pyrrole did not offer the desired product 24.
The stereoselectivity of the reaction was further demonstrated. Several typical diazido indolines (3, 4, 10, 12) were selected for chiral HPLC analysis and two peaks (1:1) were observed, indicating that they are stereoisomers (see the Supporting information). The single trans-stereoisomer of 10 was further unequivocally confirmed by 2D NMR, in which there is no significant NOE observed between Ha (CH) and Hb (CH3) (Supporting information). Taken together, these results demonstrate that this methodology produces only the trans-stereoisomer of diazido indolines (3-21, 23). Base on these results, it is not surprising that 2-pyrimidyl substituted tryptophan gave 19 in a 1:1 ratio of diastereomers, due to the chiral carbon in tryptophan.
Then, the efficiency of the different directing groups was studied. The 2-pyridyl group gave the trans-stereoisomers of 2, 3-diazido indolines 25-32 in satisfying yields (Scheme 3). Similar to the 2-pyrimidyl group, electron-donating groups, including methyl (26, 27, 30), gave higher yields than electron-withdrawing groups (28, 29, 31, 32). Scheme 3 also shows that 2-picolinamide was an efficient directing group, and the corresponding products 33-37 were obtained in 63%-88% yield. It is most likely that Cu(OA)2 coordinated to the 2-picolinamide group of indole and formed a six-membered ring structure, leading to product formation. Additionally, 7-azaindole with an added pyridine group gives a moderate yield of the corresponding product 38 in 65% yield.

|
Download:
|
| Scheme 3. Scope of the directing group. Reaction conditions: 1 (0.3 mmol), 2 (0.9 mmol), Cu(OAc)2·H2O (0.06 mmol), PhI(OAc)2 (0.9 mmol), aqueous solution 2.5 mL (contained 20% acetone) at 25 ℃ for 40 min. Isolated yields. | |
{kind=link}
To examine the gram-scale applicability of this directing groupassisted dearomatization of indole, a reaction using 1a (6 mmol) as the substrate was performed under the standard conditions (Scheme 4). Product 3 was obtained in 88% yield as a single trans-stereomer in the absence of any ligand.

|
Download:
|
| Scheme 4. Gram scale synthesis. | |
{kind=link}
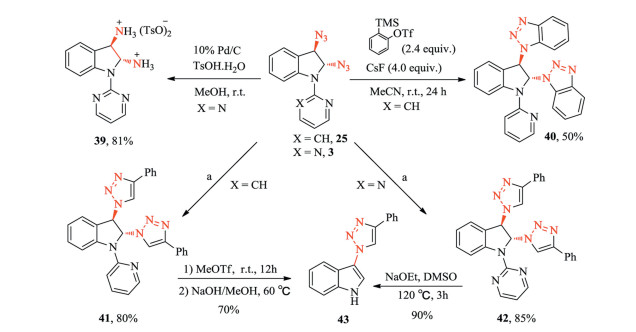
Subsequently, the synthetic utility of the diazido indolines was studied. First, the hydrogenation of 3 with Pd/C conveniently produced the vicinal diamine 39 in 81% yield. Alternatively, 25 can undergo [3 + 2] cycloaddition with benzyne to deliver trans-benzotriazole indoline 40 with a reasonable yield. Then, the click reaction between trans-2, 3-diazide indolines (3, 25) and phenylacetylene were examined, yielding the desired trans-bistriazole indolines 41 and 42 in high yields (Scheme 5). After that, we tried to remove the pyridyl and pyrimidyl directing groups. The compound 41 was treated with methyltrifluoromethanesulfonate for 12 h, followed by hydrolysis with NaOH in methanol at 60 ℃ for 6 h. Surprisingly, instead of the deprotected product, the elimination product 43 was obtained in 70% yield. Similarly, the deprotection of the pyrimidyl group (42) by sodium ethoxide in DMSO did not afford the expected deprotected product, and the elimination product 43 was obtained in 90% yield.

|
Download:
|
| Scheme 5. Functionalization of diazido indolines and deprotection of the direction group. Reagents and conditions: (a) phenylacetylene (2.5 equiv.), copper sulfate pentahydrate (0.1 equiv.), L-ascorbic acid (0.2 equiv.), tBuOH/H2O = 2:1, r.t., 3 h. | |
{kind=link}
To gain insight into the mechanism of the trans-2, 3-diazidation of indoles, several control experiments were then performed. PhI(OAc)2 and TMSN3 have been reported to be involved in the generation of azido radicals [34]. Thus, the dearomatization reaction was performed in the presence of 2, 2, 6, 6-(tetramethylpiperidin-1-yl)oxyl (TEMPO), a well-known radical scavenger. Instead of the diazidation product, compound 44 was formed in 98% yield (Scheme 6a). This result demonstrated that the reaction system involves a radical process. In the presence of another radical acceptor, 1, 1-diphenylethylene, the reaction also did not offer 3, which further confirms that the reaction is a free radical reaction. In order to demonstrate the function of the directing groups (N-2-pyridyl and N-2-pyrimidyl), we designed a series of Nsubstituted indoles as control substrates.

|
Download:
|
| Scheme 6. Mechanistic studies of the diazidation of indoles. ND: no product or product less than 5%. | |
{kind=link}
As shown in Scheme 6b, 1-methyl-1H-indole (1A), 1-phenyl- 1H-indole (1B), 1-(pyridin-3-yl)-1H-indole (1C), 1-benzyl-1H-indole (1D), 1-tosyl-1H-indole (1E) and tert-butyl 1H-indole-1- carboxylate (1F) did not afford the expected diazido product and most of the starting material was recovered. These experiments indicated that the directing group could facilitate coordination with copper acetate, and thus is essential for the successful diazidation.
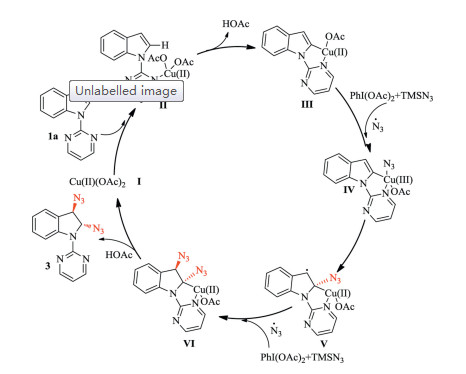
Taken together, we proposed a possible mechanism for the copper-catalyzed diazidation of indole substrates with directing groups (Scheme 7). Cu(OA)2 first coordinates to the pyrimidyl group of indole Ⅱ, forming a stable five-membered ring structure via regioselective activation of the C-H bond at the C-2 position of the indole to afford the CuⅡ complex Ⅲ, together with the loss of acetic acid. The azido radical generated by the reaction of PhI(OAc)2 and TMSN3 is transferred to the CuⅡ complex Ⅲ and coordinates to CuⅡ, forming the CuⅢ-azide complex Ⅳ. Then the azide group is rapidly inserted into the indole C-2 position. Meanwhile, the double bond in the indole C2, C3 position is dearomatized, producing the carbo radical complex Ⅴ. The trans-stereochemistry may be caused by a coplanar structure in complex Ⅴ. In order to form the stable complex Ⅵ, another azido radical is introduced to reverse side of the indoline, where it encounters less steric hindrance. Protonation of Ⅵ with acetic acid then produces product 3 and regenerates the catalyst Cu(OAc)2.

|
Download:
|
| Scheme 7. Proposed catalytic mechanism. | |
{kind=link}
In summary, we developed a copper-catalyzed 2, 3-diazidation of indole derivatives via C-H activation using azidotrimethylsilane as the azide source. The methodology provides the synthesis of a variety of 2, 3-diazide-substituted indoles in high yields. The reaction exhibits broad substrate tolerance and high diastereoselectivity under relatively mild conditions. Moreover, the indole derivatives react well in aqueous solution with no need for inert gas atmosphere. To the best of our knowledge, the reaction mechanism represents the first example of pyrimidyl, pyridyl and picolinamide as directing groups for the direct diazidation of indoles via C-H activation. Furthermore, the azide group can be easily converted into other functional groups, including vicinal diamines, triazoles and benzotriazoles. This gram-scale methodology has wide applicability for the synthesis of complex molecules or natural products containing indoline moieties.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors are grateful to the National Natural Science Foundation of China (Nos. 21472172, 21272212), Natural Science Foundation of Zhejiang Province (No. LY17B060009).
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.035.
| [1] |
Y. Matsuda, M. Kitajima, H. Takayama, Org. Lett. 10 (2008) 125-128. DOI:10.1021/ol702637r |
| [2] |
Q. Li, T.T. Xia, L.C. Yao, H.T. Deng, X.B. Liao, Chem. Sci. 6 (2015) 3599-3605. DOI:10.1039/C5SC00338E |
| [3] |
M.H. Wang, Y.C. Hu, B.D. Sun, et al., Org. Lett. 20 (2018) 1806-1809. DOI:10.1021/acs.orglett.8b00304 |
| [4] |
C. Liu, J.C. Yi, Z.B. Zheng, et al., Angew. Chem. Int. Ed. 55 (2016) 751-754. DOI:10.1002/anie.201508570 |
| [5] |
Y.A. Yuan, D.F. Lu, Y.R. Chen, H. Xu, Angew. Chem. Int. Ed. 55 (2016) 534-538. DOI:10.1002/anie.201507550 |
| [6] |
T.R. Reddy, D.S. Rao, S. Kashyap, Chem. Commun. 55 (2019) 2833-2836. DOI:10.1039/C9CC00007K |
| [7] |
H.H. Peng, Z.L. Yuan, P.H. Chen, G.S. Liu, Chin. J. Chem. 35 (2017) 876-880. DOI:10.1002/cjoc.201600834 |
| [8] |
N. Fu, G.S. Sauer, A. Saha, A. Loo, S. Lin, Science 357 (2017) 575-579. DOI:10.1126/science.aan6206 |
| [9] |
D. Lubriks, I. Sokolovs, E. Suna, J. Am. Chem. Soc. 134 (2012) 15436-15442. DOI:10.1021/ja305574k |
| [10] |
C.S. Azad, A.K. Narula, RSC Adv. 5 (2015) 100223-100227. DOI:10.1039/C5RA21963A |
| [11] |
S.N. Thibault, A. Rayar, P. Retailleau, K. Cariou, R.H. Dodd, Chem. -Eur. J. 21 (2015) 14205-14210. DOI:10.1002/chem.201501782 |
| [12] |
Y.D. Dou, X.X. Gu, S.S. Ying, et al., Org. Biomol. Chem. 16 (2018) 712-716. DOI:10.1039/C7OB02881D |
| [13] |
M. Gehringer, S.A. Laufer, Angew. Chem. Int. Ed. 56 (2017) 2-4. DOI:10.1002/anie.201611453 |
| [14] |
J. Ohata, Y. Zeng, L. Segatori, Z.T. Ball, Angew. Chem. Int. Ed. 57 (2018) 4015-4019. DOI:10.1002/anie.201800828 |
| [15] |
Y. Tamura, S. Kwon, F. Tabusa, M. Ikeda, Tetrahedron Lett. 38 (1975) 3291-3294. DOI:10.1016/S0040-4039(00)91428-X |
| [16] |
R.M. Moriarty, J.S. Khosrowshahi, Tetrahedron Lett. 27 (1986) 2809-2812. DOI:10.1016/S0040-4039(00)84648-1 |
| [17] |
S.J. Shen, C.L. Zhu, D.F. Lu, H. Xu, ACS Catal. 8 (2018) 4473-4482. DOI:10.1021/acscatal.8b00821 |
| [18] |
J. Wu, Y.C. Dou, R. Guillot, C. Kouklovsky, G. Vincent, J. Am. Chem. Soc. 141 (2019) 2832-2837. DOI:10.1021/jacs.8b13371 |
| [19] |
F. Xie, Z.S. Qi, X.W. Li, Angew. Chem. Int. Ed. 52 (2013) 11862-11866. DOI:10.1002/anie.201305902 |
| [20] |
D.Q. Tu, X.Z. Cheng, Y.D. Gao, et al., Org. Biomol. Chem. 14 (2016) 7443-7446. DOI:10.1039/C6OB01281G |
| [21] |
W.T. Zeng, M. Nukeyeva, Q.M. Wang, C. Jiang, Org. Biomol. Chem. 16 (2018) 598-608. DOI:10.1039/C7OB02921G |
| [22] |
B. Sun, T. Yoshino, S. Matsunaga, M. Kanai, Adv. Synth. Catal. 356 (2014) 1491-1495. DOI:10.1002/adsc.201301110 |
| [23] |
J.J. Shi, B. Zhou, Y.X. Yang, Y.C. Li, Org. Biomol. Chem. 10 (2012) 8953-8955. DOI:10.1039/c2ob26767e |
| [24] |
K. Takamatsu, Y. Hayashi, S. Kawauchi, K. Hirano, M. Miura, ACS Catal. 9 (2019) 5336-5344. DOI:10.1021/acscatal.9b01145 |
| [25] |
F.Z. Han, S.S. Xun, L. Jia, et al., Org. Lett. 21 (2019) 5907-5911. DOI:10.1021/acs.orglett.9b02040 |
| [26] |
M.M. Lorion, N. Kaplaneris, J. Son, R. Kuniyil, L. Ackermann, Angew. Chem. Int. Ed. 58 (2019) 1684-1688. DOI:10.1002/anie.201811668 |
| [27] |
W. Chen, D.S. Ji, Y.C. Luo, Z.Y. Wang, P.F. Xu, Org. Chem. Front. 5 (2018) 1768-1771. DOI:10.1039/C8QO00232K |
| [28] |
C. Yang, G.Y. Cheng, B.F. Huang, F.T. Xue, C. Jiang, RSC Adv. 6 (2016) 87134-87141. DOI:10.1039/C6RA19741H |
| [29] |
J. Ghorai, P. Anbarasan, Org. Lett. 21 (2019) 3431-3435. DOI:10.1021/acs.orglett.9b01197 |
| [30] |
D.D. Xu, W.W. Sun, Y.L. Xie, et al., J. Org. Chem. 81 (2016) 11081-11094. DOI:10.1021/acs.joc.6b02078 |
| [31] |
L. Joucla, N. Batail, L. Djakovitch, Adv. Synth. Catal. 352 (2010) 2929-2936. DOI:10.1002/adsc.201000512 |
| [32] |
P. Nareddy, F. Jordan, M. Szostak, Org. Lett. 20 (2018) 341-344. DOI:10.1021/acs.orglett.7b03567 |
| [33] |
W.Q. Wu, S.J. Fang, G.B. Jiang, M. Li, H.F. Jiang, Org. Chem. Front. 6 (2019) 2200-2204. DOI:10.1039/C8QO01261J |
| [34] |
M. Tingoli, L. Testaferri, A. Temperini, M. Tiecco, J. Org. Chem. 61 (1996) 7085-7091. DOI:10.1021/jo960866g |