 2020, Vol. 31
2020, Vol. 31 


b Beijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
Aliphatic amines are an important class of structural motifs ubiquitously found in pharmaceuticals and agrochemicals [1]. Among various approaches for amine synthesis [2], direct C(sp3)-H functionalization of readily available amines provides one of the most efficient and straightforward strategies for efficient construction of complex amine targets. However, the existing methods on remote C(sp3)-H functionalization [3-6] often require pre-installation of various protecting groups or directing auxiliaries to improve the stability and reactivity, and the removal of these groups after functionalization is also needed to release the functionalized amines. Although being atom- and step-economical, native amine-directed C(sp3)-H functionzalization of aliphatic amines has been far less explored [7]. Successful examples were mainly restricted to β- and γ-C(sp3)-H functionalization of sterically bulky secondary amines [8]. Thus far, NH2-directed remote C(sp3)-H functionalization of primary aliphatic amines has been a long-standing challenge in the past decade. The main reason for this limitation might be that primary amines have a higher propensity to forming less active bis-amine/ metal complexes with metal catalysts than secondary and tertiary amines [9]. To overcome this limitation, Shi adopted a protonation strategy [10] by using AcOH as the solvent and realized the first palladium-catalyzed γ-C(sp3)-H acetoxylation of primary aliphatic amines with PhI(OAc)2 [11].
Despite the pioneering work by Shi, efforts on NH2-directed remote C(sp3)-H arylation of primary aliphatic amines met with unsatisfactory results. According to the reports on transient ligand enabled γ- and δ-C(sp3)-H arylation of primary aliphatic amines by Dong [12], Yu [13], Ge [14] and others [15-20], transient ligands were very crucial for the high yield of these reactions (reaction 1, Scheme 1). Therefore, the challenge in the NH2-directed remote C(sp3)-H arylation of aliphatic amines was not conquered until two recent reports. One was the Pd-catalyzed γ-C(sp3)-H arylation of α, α-dialkyl-α-amino esters with diaryliodonium triflates developed by our group (reaction 2, Scheme 1) [21]. However, further applying this protocol to the C-H arylation of other primary aliphatic amines without α-CO2R (for example, tert-amylamine) led to poor yield (< 40%). In addition, the harsh conditions in the preparation of diaryliodonium triflates also limited the broad application of this protocol. Another report was the Pd-catalyzed o-nitrobenzoic acidpromoted δ-C(sp3)-H and C(sp2)-H arylation with aryl iodides developed by Bannister and his co-workers [22] (reaction 3, Scheme 1). Although two examples on the γ-C(sp3)-H arylation were also provided, theyield was verylow (38% and48%). Therefore, development of new protocols for efficient γ-C(sp3)-H arylation o primary aliphatic amines with easily available aryl iodides is very desirable. In this context and in continuation with our previous work, we developed and herein reported a simple but efficient method for palladium-catalyzed site-selective C(sp3)-H arylation of primary aliphatic amines with aryl iodides (reaction 4, Scheme 1). A series of α-branched primary aliphatic amines including β-/γ-amino ethers and silylethers were arylated efficiently at the γ- or δ-positions [23] by a wide range of aryl iodides. And the synthetic application of the new method was exemplified by the synthesis of fingolimod analogue, and the conjugation with natural D-menthol and fluorescent 1, 8-naphthalimide.

|
Download:
|
| Scheme 1. Remote C(sp3)-H arylation of primary aliphatic amines. | |
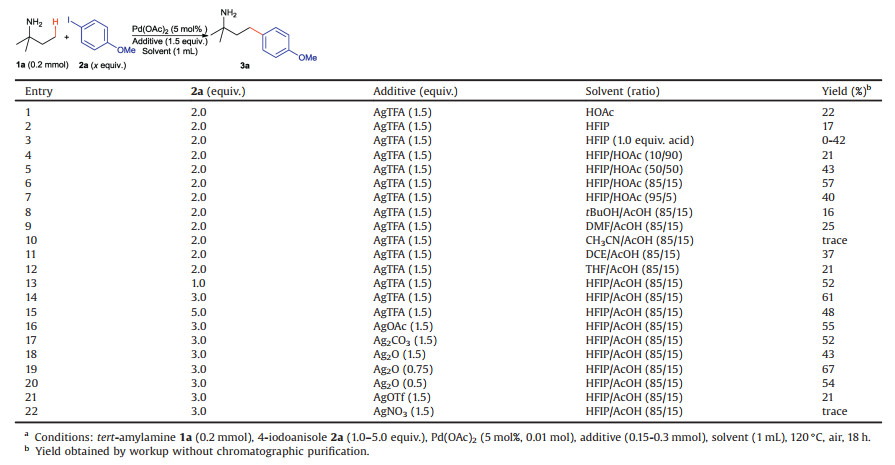
The study was commenced by conditions optimization of the reaction between tert-amylamine 1a and para-iodoanisole 2a. Initially, the treatment of 1a with 2.0 equiv. 2a, 5 mol% Pd(OAc)2, and 1.5 equiv. AgTFA in AcOH as the solvent at 120 ℃ for 18 h delivered the desired γ-arylation product 3a in 22% yield (Table 1, entry 1). However, the reaction in pure HFIP also gave the product in 17% yield (entry 2). Then, acid effect was investigated by performing the reaction in HFIP using 1.0 equiv. acid as the additive (entry 3). Among a variety of protic acids, AcOH showed strongly positive effect by improving the yield to 42%. Inspired by our previous discovery in the γ-arylation of α-amino esters by diaryliodonium triflates, we envisioned that co-solvent systems might also be beneficial to the reaction with aryl iodides. Among a variety of co-solvent systems tested, the combination of HFIP/ AcOH in a ratio of 85/15 afforded the optimal result (entry 6). Either changing the ratios of HFIP/AcOH (entries 4-7) or replacing HFIP by other solvents such as tBuOH, DMF, CH3CN, dichloroethane (DCE) and THF (entries 8-12), led to reduced yield. In order to further increase the yield, a lot of silver and copper salts were tested as the additive to replace AgTFA (Table S3 in Supporting information). Silver salts with basic counteranions were all effective, among which 0.75 equiv. Ag2O gave the best result (entry 19). But the use of other silver salts having neutral counteranions resulted in much lower yield (entries 21 and 22), while all the tested copper salts showed no activity (Table S3 in Supporting information). Finally, the optimal conditions were determined as: 1a (0.2 mmol), 2a (0.6 mmol), 5 mol% Pd(OAc)2, 0.75 equiv. Ag2O, HFIP/AcOH (85/15, 1 mL), 120 ℃, air for 18 h. The reaction under these conditions gave 3a as the single product in 67% yield without chromatographic purification (entry 19).
|
|
Table 1 Conditions optimization for γ-C(sp3)-H arylation of tert-amylamine (1a) with 4-iodoanisole (2a).a |
{kind=link}
With the optimal conditions in hand, the scope of aryl iodides in the γ-arylation of tert-amylamine 1a was first investigated (Scheme 2). A large number of aryl iodides having substituents at the ortho-, meta-, and para-positions were tested. The reaction of ortho- and meta-substituted aryl iodides required longer reaction time (36 h) and/or a higher catalyst loading (10 mol%) to reach similar conversion than para-substituted counterparts (3f vs. 3r; 3e vs. 3q). Meanwhile, both electron-rich (OMe, 3a) and electron-poor aryl groups (F, 3f; CO2Me, 3l) were successfully installed to the g-position of tert-amylamine in similar yield. The result indicated that the reaction was sensitive to the steric effect, but not to the electronic property of aryl groups. Furthermore, a wide range of functional groups such as CF3, F, Cl, Br, OTs, Ac, CONHPh, CO2Me, NO2 and CN were well tolerated to the conditions. The compatibility of these functional groups not only provided useful handles for further transformations of the products, but also gave opportunities for late-stage modification of complex amines. It is worth noting that there were no N-arylation and other C-H arylation products detected in all these reactions. Therefore, chromatographic purification is not required. Besides aryl groups, a Ts-protected indole ring (3u) was also connected to the amine successfully by this transformation, which might find applications in the synthesis of indole-embedded natural products in the future.

|
Download:
|
| Scheme 2. Scope of aryl iodides for the γ-C(sp3)-H arylation of tert-amylamine (1a). Conditions: 1a (0.2 mmol), 2 (0.6 mmol), Pd(OAc)2 (5 mol%, 0.01 mol), Ag2O (0.75 equiv., 0.15 mmol), HFIP/AcOH (1 mL, 85/15, v/v), 120 ℃, air, 18 h. | |
{kind=link}
After testing the scope of aryl iodides, we continued to examine the scope of primary aliphatic amines (Scheme 3). Firstly, α-tertiary carbons were found to be indispensible for the transformation because amines having α-H such as 1p and 1q underwent facile oxidation followed by hydrolysis to afford aldehydes. A series of α, α-dialkylpropylamines were arylated at the 1° γ-carbons in moderate to good yields (4b-4f, 46%-74%). In these reactions, none of the 2° γ-C-H, benzylic C-H, 1° β-C-H and 1° δ-C-H bonds were functionalized. The excellent regioselectivity ruled out that the reaction followed a Shilov-type oxidation pathway [3], but supported that the amino group acted as the directing group for C-H functionalization. Interestingly, when there was no 1° γ-C-H bond in the substrate, 1° δ-C-H arylation could take place under the same conditions. The reaction of 2, 4, 4- trimethylpentan-2-amine with PhI provided the δ-monoarylation product 4g as the single product in 44% yield. Secondly, O-benzyl and O-silyl ethers were well compatible to the reaction conditions. The reaction of O-benzyl amino ethers afforded the γ-arylation products (4h and 4i) in moderate yields with aryl C(sp2)-H bonds and α-C(sp3)-H bonds of oxygen unaffected. Probably due to the steric effect of silyl groups, O-TBS amino ethers displayed higher reactivity than O-benzyl ethers. Two α-ethyl-α-alkyl substituted amino silylethers underwent γ-arylation to give the products 4j and 4k in much higher yields than 4i (63% and 68% vs. 44%). Also, in the reaction of α-ethyl-α-isobutyl substituted amino silylether 1m, both of γ-monoarylation and d-monoarylation products (4m) were formed in 58% total yield and 3:1 ratio. With a higher catalyst loading (10 mol%), the α-methyl-α-isobutyl amino TIPS-ether 1n was even arylated at the δ-positions to yield the mono- and diarylation products in 60% total yield and 2.3:1 ratio. The steric bulk of silyl groups also resulted in excellent diastereoselectivity. In our previous report, the γ-arylation of α-methyl-valine ester gave the product in very poor diastereoselectivity (dr 1.2:1) [21]. Surprisingly, in the reaction of valine-derived silylether 1l, only one diastereoisomer 4l was observed by 1H NMR analysis. Thirdly, the proximal COOEt might be detrimental to the reaction as the reaction of α-amino ester 1o with PhI afforded the arylation product 4o in just 16% yield. In addition, the conditions were also not suitable for secondary amines. The reaction of N-pentyl-tert-amylamine 1r did not give any C-H arylation products, but left the starting material mainly recovered.

|
Download:
|
| Scheme 3. Scope of primary aliphatic amines. Conditions: 1 (0.2 mmol), 2 (3.0 equiv.), Pd(OAc)2 (5 mol%), Ag2O (0.75 equiv.), HFIP/AcOH (1 mL, 85/15, v/v), 120 ℃, air, 18 h. | |
{kind=link}
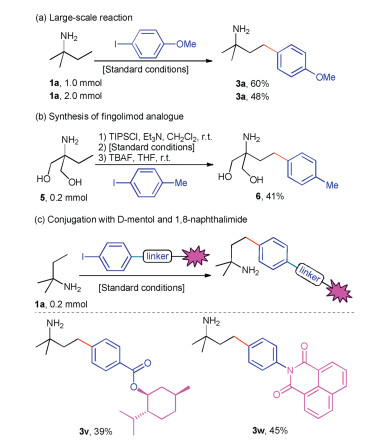
Finally, the synthetic utility of the new protocol was explored. First, the γ-arylation reaction of 1a was conducted at 5 or 10 times the scale for the optimal conditions and delivered the product 3a in 60% and 48% yield (Scheme 4a). Furthermore, using the γ-arylation reaction as the key step, a fingolimod analogue 6 was also prepared from the commercial2-amino-2-ethylpropane-1, 3-diol in 41% yield via a three-step synthesis (Scheme 4b). In addition, with ester and imide groups as the linkers, natural D-menthol and fluorescent 1, 8- naphthalimide were connected to the aryl moiety of PhI. Applying the conditions to the reaction of tert-amylamine with the two modified aryl iodides resulted in the formation of two conjugation products 3v and 3w in 39% and 45% yield respectively (Scheme 4c).

|
Download:
|
| Scheme 4. Application of the method. | |
{kind=link}
In conclusion, we have developed a simple but efficient protocol for site-selective arylation of primary aliphatic amines by aryl iodides. With only 5 mol% Pd(OAc)2, a wide range of primary amines including β-/γ-amino ethers were efficiently arylated at the γ- or δ-C-H bonds by a large number of aryl iodides with diverse functional groups. The synthetic utility of this method was also explored by the conjugation of tert-amylamine with natural D-menthol and fluorescent 1, 8-naphthalimide, and the synthesis of a fingolimod analogue.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe thank the National Natural Science Foundation of China (No. 21502006), Beijing National Laboratory for Molecular Sciences and Beijing Institute of Technology (BIT) for financial support. The Analysis and Testing Centre of Beijing Institute of Technology is highly appreciated for their instrument support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2019.10.034.
| [1] |
(a) S. Jeanmart, A.J.F. Edmunds, C. Lamberth, M. Pouliot, Bioorg. Med. Chem. 24 (2016) 317-341; (b) P. Das, M.D. Delost, M.H. Qureshi, D.T. Smith, J.T. Njardarson, J. Med. Chem. 62 (2019) 4265-4311; (c) F.Z. Dörwald, Aliphatic amines, in: F.Z. Dörwald (Ed.), Lead Optimization for Medicinal Chemists: Pharmacokinetic Properties of Functional Groups and Organic Compounds, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2012, pp. 107-117. |
| [2] |
(a) J.F. Knifton, Catal. Today 36 (1997) 305-310; (b) Y. Park, Y. Kim, S. Chang, Chem. Rev. 117 (2017) 9247-9301; (c) P. Kalck, M. Urrutigoity, Chem. Rev. 118 (2018) 3833-3861. |
| [3] |
(a) B.D. Dangel, J.A. Johnson, D. Sames, J. Am. Chem. Soc. 123 (2001) 8149-8150; (b) M. Lee, M.S. Sanford, J. Am. Chem. Soc. 137 (2015) 12796-12799; (c) C.T. Mbofana, E. Chong, J. Lawniczak, M.S. Sanford, Org. Lett. 18 (2016) 4258-4261; (d) M. Lee, M.S. Sanford, Org. Lett. 19 (2017) 572-575. |
| [4] |
(a) C. Martínez, K. Muñiz, Angew. Chem. Int. Ed. 54 (2015) 8287-8291; (b) E.A. Wappes, S.C. Fosu, T.C. Chopko, D.A. Nagib, Angew. Chem. Int. Ed. 55 (2016) 9974-9978; (c) G.J. Choi, Q. Zhu, D.C. Miller, C.J. Gu, R.R. Knowles, Nature 539 (2016) 268; (d) J.C.K. Chu, T. Rovis, Nature 539 (2016) 272; (e) M.Y. Yang, B. Su, Y. Wang, et al., Nat. Commun. 5(2014) 5707; (f) Z. Li, Q. Wang, J. Zhu, Angew. Chem. Int. Ed. 57 (2018) 13288-13292. |
| [5] |
S. Munnuri, A.M. Adebesin, M.P. Paudyal, et al., J. Am. Chem. Soc. 139 (2017) 18288-18294. DOI:10.1021/jacs.7b09901 |
| [6] |
(a) G. Rouquet, N. Chatani, Angew. Chem. Int. Ed. 52 (2013) 11726-11743; (b) O. Daugulis, J. Roane, L.D. Tran, Acc. Chem. Res. 48 (2015) 1053-1064; (c) B. Liu, Y.Q. Han, F. Hu, B.F. Shi, Palladium-catalyzed directed arylation of unactivated C(sp3)-H bonds, in: A.R. Kapdi, D. Maiti (Eds.), Strategies for Palladium-Catalyzed Non-Directed and Directed C-H Bond Functionalization, Elsevier, 2017, pp. 167-203; (d) C. He, W.G. Whitehurst, M.J. Gaunt, Chem. 5(2019) 1031-1058. |
| [7] |
(a) Z. Liang, L. Ju, Y. Xie, L. Huang, Y. Zhang, Chem. -Eur. J. 18 (2012) 15816-15821; (b) C. Suzuki, K. Hirano, T. Satoh, M. Miura, Org. Lett. 15 (2013) 3990-3993; (c) A. Boelke, L.D. Caspers, B.J. Nachtsheim, Org. Lett. 19 (2017) 5344-5347; (d) K. Orito, A. Horibata, T. Nakamura, et al., J. Am. Chem. Soc. 126 (2004) 14342-14343; (e) A. Lazareva, O. Daugulis, Org. Lett. 8(2006) 5211-5213; (f) B. Haffemayer, M. Gulias, M.J. Gaunt, Chem. Sci. 2(2011) 312-315; (g) M. Miura, C.G. Feng, S. Ma, J.Q. Yu, Org. Lett. 15 (2013) 5258-5261; (h) A. Mancinelli, C. Alamillo, J. Albert, et al., Organometallics 36 (2017) 911-919; (i) C.H. Zhang, Y.Z. Ding, Y.Z. Gao, S.D. Li, G. Li, Org. Lett. 20 (2018) 2595-2598. |
| [8] |
(a) A. McNally, B. Haffemayer, B.S.L. Collins, M.J. Gaunt, Nature 510 (2014) 129-133; (b) J. Calleja, D. Pla, T.W. Gorman, et al., Nat. Chem. 7(2015) 1009-1016; (c) C. He, M.J. Gaunt, Angew. Chem. Int. Ed. 54 (2015) 15840-15844; (d) D. Willcox, B.G.N. Chappell, K.F. Hogg, et al., Science 354 (2016) 851. |
| [9] |
A.D. Ryabov, Chem. Rev. 90 (1990) 403-424. DOI:10.1021/cr00100a004 |
| [10] |
A.P. Smalley, M.J. Gaunt, J. Am. Chem. Soc. 137 (2015) 10632-10641. DOI:10.1021/jacs.5b05529 |
| [11] |
K. Chen, D. Wang, Z.W. Li, et al., Org. Chem. Front. 4 (2017) 2097-2101. DOI:10.1039/C7QO00432J |
| [12] |
Y. Xu, M.C. Young, C. Wang, D.M. Magness, G. Dong, Angew. Chem. Int. Ed. 55 (2016) 9084-9087. DOI:10.1002/anie.201604268 |
| [13] |
(a) Y. Wu, Y.Q. Chen, T. Liu, M.D. Eastgate, J.Q. Yu, J. Am. Chem. Soc. 138 (2016) 14554-14557; (b) Y.Q. Chen, Z. Wang, Y. Wu, et al., J. Am. Chem. Soc.140 (2018) 17884-17894. |
| [14] |
Y. Liu, H. Ge, Nat. Chem. 9 (2017) 26-32. DOI:10.1038/nchem.2606 |
| [15] |
A. Yada, W. Liao, Y. Sato, M. Murakami, Angew. Chem. Int. Ed. 56 (2017) 1073-1076. DOI:10.1002/anie.201610666 |
| [16] |
M. Kapoor, D. Liu, M.C. Young, J. Am. Chem. Soc. 140 (2018) 6818-6822. DOI:10.1021/jacs.8b05061 |
| [17] |
H. Lin, C. Wang, T.D. Bannister, T.M. Kamenecka, Chem. -Eur. J. 24 (2018) 9535-9541. DOI:10.1002/chem.201802465 |
| [18] |
John-Campbell S. St, A.K. Ou, J.A. Bull, Chem. -Eur. J. 24 (2018) 17838-17843. DOI:10.1002/chem.201804515 |
| [19] |
J.T. Ye, I. Kalvet, F. Schoenebeck, T. Rovis, Nat. Chem. 10 (2018) 1037-1041. DOI:10.1038/s41557-018-0085-9 |
| [20] |
X.X. Hu, J.B. Liu, L.L. Wang, et al., Org. Chem. Front. 5 (2018) 1670-1678. DOI:10.1039/C8QO00094H |
| [21] |
P.K. Pramanick, Z. Zhou, Z.L. Hou, B. Yao, J. Org. Chem. 84 (2019) 5684-5694. DOI:10.1021/acs.joc.9b00605 |
| [22] |
H. Lin, X. Pan, A.L. Barsamian, T.M. Kamenecka, T.D. Bannister, ACS Catal. 9 (2019) 4887-4891. DOI:10.1021/acscatal.8b04927 |
| [23] |
(a) S. Guin, P. Dolui, X. Zhang, et al., Angew. Chem. Int. Ed. 58 (2019) 5633-5638; (b) B.B. Zhan, Y. Li, J.W. Xu, et al., Angew. Chem. Int. Ed. 57 (2018) 5858-5862; (c) J.W. Xu, Z.Z. Zhang, W.H. Rao, B.F. Shi, J. Am. Chem. Soc. 138 (2016) 10750-10753. |