 2020, Vol. 31
2020, Vol. 31 


b College of Chemistry and Materials Engineering, Wenzhou University, Wenzhou 325035, China;
c State Key Laboratory of Organometallic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Since the last decade, gold-catalyzed nucleophilic addition reactions to alkynes have received tremendous interest and been widely used in the facile synthesis of an incredible variety of complex molecules, especially the valuable cyclic compounds [1]. Among these, the terminal alkynes can undergo various highly regioselective transformations catalyzed by gold, thus providing an efficient way for C—C, C—H and C—X bond formation. However, Markovnikov regioselectivity is normally observed in this kind of nucleophilic addition (Scheme 1a), and recent reports involving the anti-Markovnikov regioselectivity are mostly limited to reactions driven by aromatization [2a–c] and involving the gold vinylidene intermediate [2d, e]. Of note, the dominance of the LUMO coefficient on the internal carbon in the π-complexes of terminal alkynes and gold(Ⅰ) complexes has been computed in detail, even involving a fully relativistic treatment [3]. Recently, our group has achieved a variety of gold-catalyzed antiMarkovnikov cycloisomerization-initiated cascade cyclizations by utilizing the steric strain in ring formation [4h], and this strategy has been applied to the synthesis of various N-heterocycles from readily available homopropargyl alcohols or amides [4]. Inspired by these results and our recent study on the coppercatalyzed tandem reaction of sulfonyl-protected indole tethered homopropargyl amides [4g], we envisioned that the gold antiMarkovnikov cycloisomerization might be realized by choosing the suitable protecting groups (Scheme 1b) [5]. However, it remains a highly challenging task because of the difficulty in preventing the competing Markovnikov cyclization [6] and achieving the desired cascade cyclization [7].

|
Download:
|
| Scheme 1. Gold-catalyzed nucleophilic addition to terminal alkynes. | |
Herein, we disclose a gold-catalyzed anti-Markovnikov cycloisomerization-initiated tandem reaction by employing the Bocprotected indole tethered homopropargyl amides as substrates [8, 9], enabling facile access to a wide range of valuable bridged aza-[n.2.1] skeletons (n = 3–7) at room temperature with high diastereoselectivity and enantioselectivity by a chirality-transfer strategy.
At the outset, Boc-protected indole tethered chiral homopropargyl amide 1a was chosen as the model substrate (Table 1). To our delight, typical gold catalysts such as Ph3PAuNTf2 and IPrAuNTf2 could indeed catalyze the cascade cyclization reaction to produce the desired aza-[3.2.1] skeleton 2a, albeit along with the byproduct 2aa, which was formed presumably through a direct gold-catalyzed Markovnikov cycloisomerization-initiated tandem reaction (entries 1 and 2; Supporting information for details). We then investigated other gold catalysts and were delighted to find that excellent yields were achieved by employing Cy-JohnPhosAuNTf2 and BrettPhosAuNTf2 as catalysts (entries 3 and 4). Of note, by employing PtCl2 as the catalyst, the occurrence of 2aa could become dominant (entry 5). In contrast to the gold catalysts, other transition metal catalysts and Brønsted acids promoted selective formation of 2ab (entries 6–10) [10]. Finally, it was found that the reaction proceeded equally well in DCM as the solvent (entry 11).
|
|
Table 1 Optimization of reaction conditions. |

{kind=link}
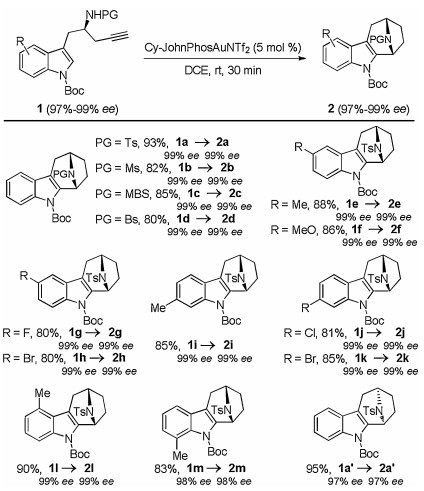
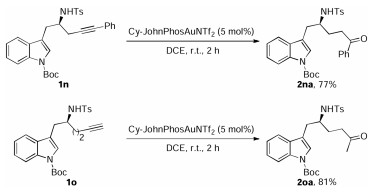
With the optimized reaction conditions in hand, the scope of this gold-catalyzed tandem reaction was explored, as shown in Scheme 2. Chiral Boc-protected indole tethered homopropargyl amides 1 were accessed with excellent enantiomeric excesses (97%–99% ee) from readily available indolyl aldehydes by Ellman's tert-butylsulfinamide chemistry (Supporting information for details). Different N-protecting groups were first screened, and it was found that the reaction occurred efficiently to afford the corresponding aza-[3.2.1] skeletons 2a–d in 80%–93% yields (2a was confirmed by X-ray diffraction analyses (CCDC No. 1554636)). Additionally, the reaction was also extended to amides bearing different substituents on the indole ring, leading to the desired products 2e–k in high yields. Notably, the reaction could be even extended to sterically hindered indolyl substrates 1l and 1m, and the desired products 2l and 2m were formed in high yields. Our attempts to extend the reaction to internal alkyne 1n and indolyl amide 1o only resulted in the formation of the corresponding hydration products 2na (77%) and 2oa (81%), respectively (Scheme 3). Finally, the reaction also proceeded smoothly with (S)-(+)-tert-butylsulfinamide-derived 1a', delivering the desired 2a' with the opposite enantioselectivity. Importantly, complete chirality transfer was observed in all cases.

|
Download:
|
| Scheme 2. Synthesis of bridged aza-[3.2.1] skeletons 2. Reactions run in vials; [1] = 0.1 mol/L, isolated yields are reported. | |
{kind=link}

|
Download:
|
| Scheme 3. Gold-catalyzed cascade cyclization of homopropargyl amides 1n and 1o. | |
{kind=link}
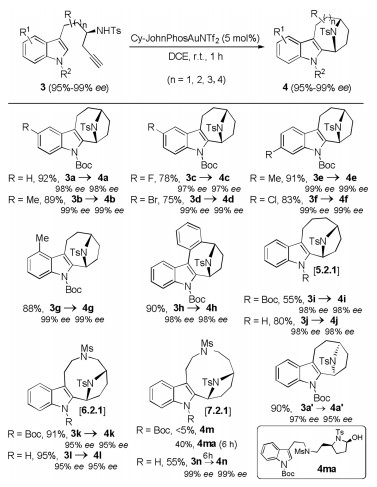
Moreover, this gold-catalyzed tandem reaction was also extended to the efficient synthesis of other bridged aza-[n.2.1] skeletons (Scheme 4). Various chiral indolyl homopropargyl amides 3 were suitable substrates for this tandem cyclization to furnish the desired bridged aza-[n.2.1] skeletons 4 in mostly good to excellent yields. A variety of amides containing various substituents on the indole ring were readily tolerated, leading to the corresponding aza-[4.2.1] skeletons 4a–g in good to excellent yields. In addition, the reaction worked satisfactorily with aryllinked substrate 3h, thus providing the desired 4h in 90% yield. (S)-(+)-tert-Butylsulfinamide-derived 3a' were also compatible with this gold catalysis, and the desired 4a' with the opposite enantioselectivity was formed in 90% yield. Finally, it was found that this tandem reaction was viable for the construction of the aza-[n.2.1] skeletons 4i–n (n = 5–7) in 55%–95% yields except for substrate 3m, and significantly improved yields were achieved when unprotected indolyl homopropargyl amides were used (4k was confirmed by X-ray diffraction analyses (CCDC No. 1554638)). While five- and six-membered rings are formed in most goldcatalyzed intramolecular reactions, such medium-sized rings are very rare in gold catalysis [11, 6b, 6c]. Again, complete chirality transfer was observed in all cases.

|
Download:
|
| Scheme 4. Synthesis of bridged aza-[3.2.1] skeletons 4. Reactions run in vials; [3] = 0.1 mol/L, isolated yields are reported. | |
{kind=link}
Interestingly, we were delighted to find that this gold-catalyzed cascade cyclization could be further extended to the homopropargyl alcohol. As shown in Scheme 5, the tandem reaction of homopropargyl alcohol 5 under the gold-catalyzed standard conditions led to the bridged oxa-[3.2.1] skeleton 6 in 62% yield. Notably, the reaction failed to give even a trace of 6 under the previous copper catalysis [4g]. A likely reason is that the homopropargyl alcohol substrate, which is not as stable as the homopropargyl amide substrates, would undergo decomposition easily at high temperature (80 ℃) required in the coppercatalyzed protocol.

|
Download:
|
| Scheme 5. Gold-catalyzed cascade cyclization of homopropargyl alcohol 5. | |
{kind=link}
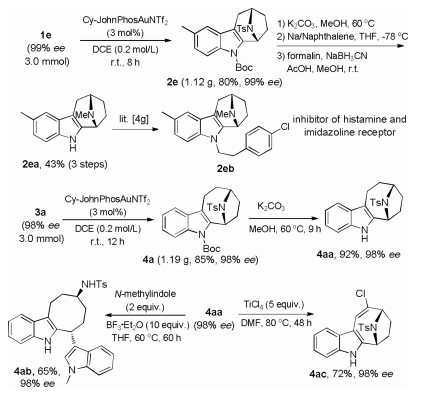
Further synthetic transformations of the above products were then explored (Scheme 6). The N–Ts group in 2e, prepared on a gram scale in 80% yield, could be readily converted into the desired N–Me group, delivering 2ea in 43% yield (3 steps), which underwent facile alkylation to eventually produce the bioactive 2eb [4g]. In addition, deprotection of 4a (gram scale: 85% yield) afforded 4aa, which could be further transformed into the valuable indole-fused cyclooctane 4ab [12] and chlorinated aza-[4.2.1] skeleton 4ac (4ac was confirmed by X-ray diffraction analyses (CCDC No. 1566099)). Importantly, enantioselectivity was well maintained in all these cases.

|
Download:
|
| Scheme 6. Gram scale reaction and synthetic applications. | |
{kind=link}
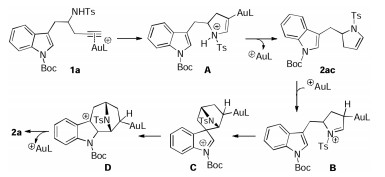
To probe the reaction mechanism, we first tried to trap the likely reaction intermediate (Scheme 7a). To our delight, the indolyl dihydropyrrole 2ac was isolated in 43% yield when the reaction ran at 0 ℃. Moreover, 2ac could be readily converted into the desired 2a in the presence of gold catalyst while no 2a was observed without gold catalyst. These results strongly support that 2ac is the key intermediate for this tandem reaction [5], and gold catalyzes both the hydroamination and Friedel℃rafts alkylation process. In addition, a deuterium labeling study revealed that almost no deuterium loss was observed (Scheme 7b), thus ruling out the gold vinylidene intermediate pathway.

|
Download:
|
| Scheme 7. Control experiments. | |
{kind=link}
On the basis of the above experimental observations and previously published results [4], a rationale for the formation of the bridged heterocycle 2a is shown in Scheme 8. The reaction starts with formation of the vinyl gold intermediate A by alkyne π coordination and concomitant 5-endo-dig cyclization. Intermediate A then undergo facile protodeauration, leading to indolyl dihydropyrrole 2ac, which could be further converted into iminium species B catalyzed by gold. Subsequent Friedel-rafts alkylation at C-3 of the indole followed by a 1, 2-migration [6g], and aromatization/protodeauration process delivers the final product 2a along with the regeneration of the gold catalyst.

|
Download:
|
| Scheme 8. Plausible mechanism. | |
{kind=link}
In summary, we have developed a gold-catalyzed antiMarkovnikov cycloisomerization-initiated cascade cyclization of Boc-protected indole tethered homopropargyl amides, delivering a wide range of bridged aza-[n.2.1] skeletons at room temperature with high diastereoselectivity and enantioselectivity by a chiralitytransfer strategy. Moreover, the gold-catalyzed tandem reaction of homopropargyl alcohol is also achieved to produce the bridged oxa-[3.2.1] skeleton. In addition, the mechanistic rationale for this cascade cyclization is well supported by a variety of control experiments, and thus the mechanism of this gold catalysis is distinctively different from the previous copper catalysis [4g].
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (Nos. 21622204, 21572186, 21772161, 21572163 and 21372178), the Natural Science Foundation of Fujian Province of China (No. 2019J02001), the President Research Funds from Xiamen University (No. 20720180036), NFFTBS (No. J1310024), PCSIRT, and Science & Technology Cooperation Program of Xiamen (No. 3502Z20183015).
Appendix A. Supplementary dataSupplementarymaterial related to this article canbefound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.019.
| [1] |
(a) E. Aguilar, J. Santamaría, Org. Chem. Front. 6 (2019) 1513-1540; (b) L. Li, T.D. Tan, Y.Q. Zhang, X. Liu, L.W. Ye, Org. Biomol. Chem.15 (2017) 8483-8492; (c) W. Zi, F.D. Toste, Chem. Soc. Rev. 45 (2016) 4567-4589; (d) A.M. Asiri, A.S.K. Hashmi, Chem. Soc. Rev. 45 (2016) 4471-4503; (e) Z. Zheng, Z. Wang, Y. Wang, L. Zhang, Chem. Soc. Rev. 45 (2016) 4448-4458; (f) D. Pflästerer, A.S.K. Hashmi, Chem. Soc. Rev. 45 (2016) 1331-1367; (g) D.B. Huple, S. Ghorpade, R. Liu, Adv. Synth. Catal. 358 (2016) 1348-1367; (h) L. Liu, J. Zhang, Chem. Soc. Rev. 45 (2016) 506-516; (i) F. Pan, C. Shu, L.W. Ye, Org. Biomol. Chem. 14 (2016) 9456-9465; (j) D. Qian, J. Zhang, Chem. Soc. Rev. 44 (2015) 677-698; (k) R. Dorel, A.M. Echavarren, Chem. Rev. 115 (2015) 9028-9072; (l) Y. Yamamoto, Chem. Soc. Rev. 43 (2014) 1575-1600; (m) H.S. Yeom, S. Shin, Acc. Chem. Res. 47 (2014) 966-977; (n) L. Fensterbank, M. Malacria, Acc. Chem. Res. 47 (2014) 953-965; (o) C. Obradors, A.M. Echavarren, Acc. Chem. Res. 47 (2014) 902-912; (p) Y.M. Wang, A.D. Lackner, F.D. Toste, Acc. Chem. Res. 47 (2014) 889-901; (q) L. Zhang, Acc. Chem. Res. 47 (2014) 877-888; (r) A.S.K. Hashmi, Acc. Chem. Res. 47 (2014) 864-876. |
| [2] |
(a) A. Gimeno, A.B. Cuenca, S. Suárez-Pantiga, et al., Chem. -Eur. J. 20 (2014) 683-688; (b) R.S. Menon, A.D. Findlay, A.C. Bissember, M.G. Banwell, J. Org. Chem. 74 (2009) 8901-8903; (c) V. Mamane, P. Hannen, A. Fürstner, Chem. -Eur. J. 10 (2004) 4556-4575; (d) D.J. Ye, J.F. Wang, X. Zhang, et al., Green Chem. 11 (2009) 1201-1208; (e) I.V. Seregin, V. Gevorgyan, J. Am. Chem. Soc. 128 (2006) 12050-12051. |
| [3] |
M. Pernpointner, A.S.K. Hashmi, J. Chem. Theory Comput. 5 (2009) 2717-2725.
|
| [4] |
(a) C. Shu, M.Q. Liu, Y.Z. Sun, L.W. Ye, Org. Lett. 14 (2012) 4958-4961; (b) C. Shu, M.Q. Liu, S.S. Wang, L. Li, L.W. Ye, J. Org. Chem. 78 (2013) 3292-3299; (c) Y.F. Yu, C. Shu, B. Zhou, et al., Chem. Commun. (Camb.) 51 (2015) 2126-2129; (d) C. Shu, L. Li, C.H. Shen, et al., Chem. -Eur. J. 22 (2016) 2282-2290; (e) Y.F. Yu, C. Shu, T.D. Tan, et al., Org. Lett. 18 (2016) 5178-5181; (f) T.D. Tan, Y.B. Chen, M.Y. Yang, et al., Chem. Commun. (Camb.) 5 (2019) 9923-9926; (g) T.D. Tan, X.Q. Zhu, H.Z. Bu, et al., Angew. Chem. Int. Ed. 58 (2019) 9632-9639; (h) C. Shu, L. Li, T.D. Tan, D.Q. Yuan, L.W. Ye, Sci. Bull. (Beijing) 62 (2017) 352-357. |
| [5] |
R. Ali, G. Singh, S. Singh, R.S. Ampapathi, W. Haq, Org. Lett. 18 (2016) 2848-2851.
|
| [6] |
(a) L. Zhang, Y. Wang, Z.J. Yao, S. Wang, Z.X. Yu, J. Am. Chem. Soc. 137 (2015) 13290-13300; (b) D. Pflästerer, S. Schumacher, M. Rudolph, A.S.K. Hashmi, Chem. -Eur. J. 21 (2015) 11585-11589; (c) D. Pflästerer, E. Rettenmeier, S. Schneider, et al., Chem. -Eur. J. 20 (2014) 6752-6755; (d) Z. Dong, C.H. Liu, Y. Wang, M. Lin, Z.X. Yu, Angew. Chem. Int. Ed. 52 (2013) 14157-14161; (e) L. Huang, H.B. Yang, D.H. Zhang, et al., Angew. Chem. Int. Ed. 52 (2013) 6767-6771; (f) L. Liu, L. Zhang, Angew. Chem. Int. Ed. 51 (2012) 7301-7304; (g) C. Ferrer, C.H.M. Amijs, A.M. Echavarren, Chem. -Eur. J. 13 (2007) 1358-1373. |
| [7] |
N. Gouault, M. Le Roch, C. Cornée, M. David, P. Uriac, J. Org. Chem. 74 (2009) 5614-5617.
|
| [8] |
(a) S. Tong, C. Piemontesi, Q. Wang, M.X. Wang, J. Zhu, Angew. Chem. Int. Ed. 56 (2017) 7958-7962; (b) T. Arto, F.J. Fañanás, F. Rodríguez, Angew. Chem.Int. Ed. 55 (2016) 7218-7221; (c) S. Hosseyni, L. Wojtas, M. Li, X. Shi, J. Am. Chem. Soc.138 (2016) 3994-3997; (d) K. Liu, C. Zhu, J. Min, et al., Angew. Chem. Int. Ed. 54 (2015) 12962-12967; (e) F.S. Zhang, Q. Lai, X.D. Shi, Z.G. Song, Chin. Chem. Lett. 30 (2019) 392-394. |
| [9] |
(a) Y. Xu, Q. Sun, T.D. Tan, et al., Angew. Chem. Int. Ed. 58 (2019) 16252-16259; (b) B. Zhou, Y.Q. Zhang, K. Zhang, et al., Nat. Commun. 10 (2019) 3234; (c) L. Li, X.Q. Zhu, Y.Q. Zhang, et al., Chem. Sci. 10 (2019) 3123-3129; (d) X.Q. Zhu, Q. Sun, Z.X. Zhang, et al., Chem. Commun. (Camb.) 54 (2018) 7435-7438; (e) X.Q. Zhu, H. Yuan, Q. Sun, et al., Green Chem. 20 (2018) 4287-4291; (f) W.B. Shen, B. Zhou, Z.X. Zhang, et al., Org. Chem. Front. 5 (2018) 2468-2472; (g) W.B. Shen, Q. Sun, L. Li, et al., Nat. Commun. 8 (2017) 1748; (h) B. Zhou, L. Li, X.Q. Zhu, et al., Angew. Chem. Int. Ed. 56 (2017) 4015-4019; (i) W.B. Shen, X.Y. Xiao, Q. Sun, et al., Angew. Chem. Int. Ed. 56 (2017) 605-609; (j) B. Zhou, Y.Q. Zhang, X. Liu, L.W. Ye, Sci. Bull. (Beijing) 62 (2017) 1201-1206; (k) C. Shu, Y.H. Wang, B. Zhou, et al., J. Am. Chem. Soc. 137 (2015) 9567-9570; (l) L. Li, B. Zhou, Y.H. Wang, et al., Angew. Chem. Int. Ed. 54 (2015) 8245-8249; (m) A.H. Zhou, Q. He, C. Shu, et al., Chem. Sci. 6 (2015) 1265-1271. |
| [10] |
M.J. James, R.E. Clubley, K.Y. Palate, et al., Org. Lett. 17 (2015) 4372-4375.
|
| [11] |
(a) D. Pflästerer, M. Rudolph, B.F. Yates, A. Ariafard, A.S.K. Hashmi, Adv. Synth. Catal. 359 (2017) 866-874; (b) T. Jime'nez, J. Carreras, J. Ceccon, A.M. Echavarren, Org. Lett.18 (2016) 1410-1413; (c) J.M. Yang, P.H. Li, Y. Wei, X.Y. Tang, M. Shi, Chem. Commun. (Camb.) 52 (2016) 346-349; (d) Y. Hu, Y. Li, S. Zhang, et al., Org. Lett. 17 (2015) 4018-4021; (e) T. Iwai, H. Okochi, H. Ito, M. Sawamura, Angew. Chem. Int. Ed. 52 (2013) 4239-4242; (f) D. Pflästerer, P. Dolbundalchok, S. Rafique, et al., Adv. Synth. Catal. 355 (2013) 1383-1393; (g) H. Ito, A. Harada, H. Ohmiya, M. Sawamura, Adv. Synth. Catal. 355 (2013) 647-652; (h) B. Bolte, F. Gagosz, J. Am. Chem. Soc. 133 (2011) 7696-7699; (i) I.D.G. Watson, S. Ritter, F.D. Toste, J. Am. Chem. Soc. 131 (2009) 2056-2057. |
| [12] |
C. Zhu, X. Zhang, X. Lian, S. Ma, Angew. Chem. Int. Ed. 51 (2012) 7817-7820.
|