 2020, Vol. 31
2020, Vol. 31 


In recent years, bridge triptycene has important applications in organic synthesis, biology, and materials. In particular, naphthacene, with its rigid scaffold, has been used as a key component in emission and viridicatumtoxin sensors [1]. Complex triptycenes are quite difficult to prepare due to their insolubility in common organic solvents [2, 3]. Diels-Alder reaction was facilitated by reagent with electron deficient structure, the acene cores could be used to achieve semiconducting materials [4]. Cycloaddition between dienophiles and anthracenes is a common method for constructing naphthacene derivatives [5]. In general, 9-10-addition and 1, 4-addition products are generally obtained by considering the high localization of π-electron at the 9, 10-position and the secondary orbital interactions. However, few reports are available on the generation of anti-1, 4-addition products as the major species under mild conditions [6]. Ma reported an AlCl3-assisted Diels– Alder reaction to obtain anti-1, 4-additions between N-maleimides and anthracene derivatives, but the 9, 10-addition product is still observed [7]. Benzynes produced by the thermal cycloisomerization of tetrayne substrates are an important reactive intermediate in organic synthesis because they have unique bond angles (sp hybridized carbon) and a benzene ring structure; therefore, the reactions of benzynes with anthracenes can generate naphthacene building blocks [8]. Herein, we demonstrated a [4 + 2] cycloaddition of hexadehydro-Diels–Alder (HDDA) derived benzyne [9] with anthracene derivatives by tuning the electronic and steric effects that generally produces either 1, 4-adducts or anticipative 9, 10- adducts as the major products with regio- and stereo-selectivity in a green, metal-free catalysis and a high atom-economic manner (Scheme 1) [10]. This protocol is compatible with a variety of sensitive functional groups and allows the synthesis of highly substituted ethenonaphthacene derivatives with multiple aromatic rings and functionalizable structures [11, 12].

|
Download:
|
| Scheme 1. Target 1, 4-adducts and formal 9, 10-adducts from HDDA-derived benzynes and anthracene derivatives. | |
We first explored the thermal cycloisomerization of 1, 3-butadiyne tethered to an alkyne group to generate a benzyne intermediate (HDDA reaction). We obtained ethenonaphthacene compounds with anthracene derivatives through this cycloaddition. With these experimental results, we optimized the reaction conditions. Anhydrous toluene was selected as the optimal solvent because the reactive benzene intermediates can easily attack residual water to generate the hydrogenation product. The product was isolated by chromatography, and the structure was confirmed by NMR analysis. In addition, the reaction is insensitive to air. The efficiency of the cascade reaction can be reasonably enhanced by increasing the reaction temperature to 100 ℃ and extending the reaction time to 8 h. Therefore, the following standard reaction conditions were chosen for subsequent experiments: 1.0 equiv. of the tetrayne substrate reacted with 1.1 equiv. of the anthracene derivative in anhydrous toluene at 100 ℃ for 8 h.
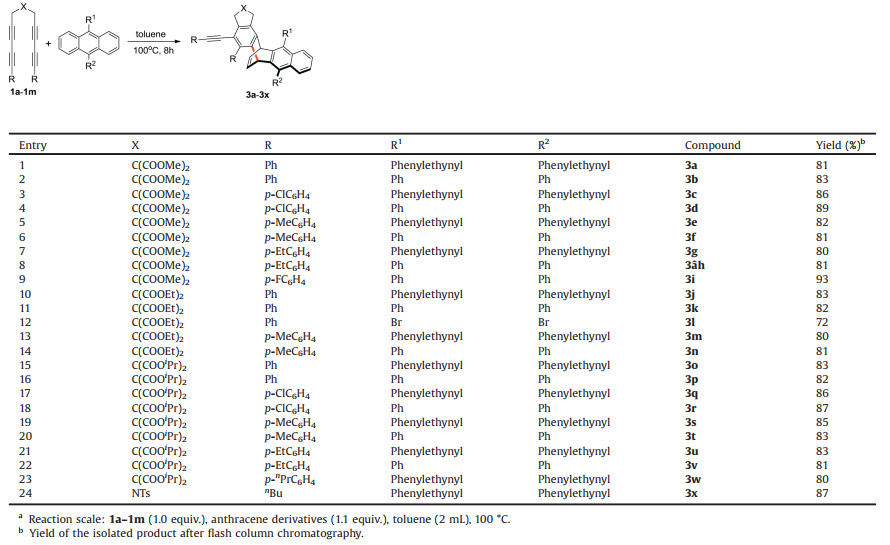
Table 1 illustrates a detailed example of the experiments conducted to expand the range of the tetrayne substrates. Products with good yield (76%–93%) could be isolated by the reaction of tetraynes containing different substituents with anthracenes. The reaction had a high tolerance for substituents on tetraynes. The electron-donating groups can participate in the reaction, and the electron-withdrawing groups displayed satisfactory performance. Product 3i showed the highest yield (93%) as pointed out from the table. Comparison of 3d with 3b, 3f, and 3h showed that the product yield was relatively high when the R-substituted group on the tetraynes was the electron-withdrawing groups. The yields of 3a-d were analyzed with the other products. The smaller the spatial structure of the substituted anthracene is, the higher the yield will be. Although 3l had the smallest spatial structure, it showed the importance of conjugated structure of the substituted anthracene in the reaction. These results indicate the potential of the addition reactions of anthracene for the synthesis of complex and highly substituted 1, 4-adducts. The configuration of the products is similar to "exo-1, 4-additon" of olefin with diene due to the reduction in the space repulsion of bulk benzynes with 9, 10- disubstituted anthracenes.
|
|
Table 1 One-pot formation of fused 1, 4-adducts.a |
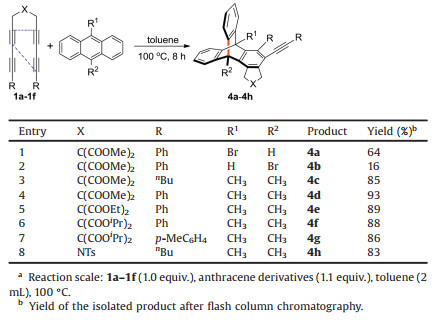
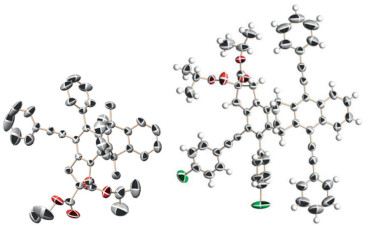
A distinct decrease in the yield of 3i was observed when using 9, 10-dibromoanthracene. Thus, we carried out a comparative experiment by using 9-bromoanthracene (Table 2). The 9, 10- adducts of 4a and 4b were obtained as a pair of diastereomer due to the desymmetrization in the presence of bromine followed by flash column chromatography. The yield is 80% at a ratio 4:1. When the substituent groups of anthracene 9, 10-position are bromine/ hydrogen, the steric hindrance is small and the 9, 10-addition occurs. When there are large substituents, such as phenyl and phenylacetylene, the 1, 4-cyclic addition occurs due to large steric hindrance. The yield of 4a is higher than that of 4b because of steric repulsion from five-membered ring on the arene with bromide. This result indicates that bulk aryne did not inhibit the addition reaction for high localization of π-electron with a single bromide at the 9-position of anthracene superior to steric effects in this case. Using methyl replacement of bromide at the 9, 10-position also obtained the corresponding products of 4d-g, with yields of 93%, 89%, 88%, and 86%, respectively. The yield of compound 4c with the aliphatic group on the terminal alkyne is 85% compared with that of 4d. The yield of 4h, which uses nitrogen instead of tethered atom of carbon, is 83%. The results demonstrate that these conversions have excellent compatibility with disubstituted tetraynes. The reaction has good regional selectivity and stereoselectivity, and the result is a single product without enantiomer because the other anthracene reagents are symmetric except 9-bromoanthracene, which have a single bromine to product diastereomer. We confirmed the molecular structure and relative configuration of compounds 3q and 4f by X-ray diffraction analysis (Fig. 1). CCDC- 1879879 (3q), CCDC-1939364 (4f) contains the supplementary crystallographic data which can be obtained free of charge from the Cambridge Crystallographic Date Cetre.
|
|
Table 2 One-pot formation of fused 9, 10-adducts.a |
{kind=link}

|
Download:
|
| Fig. 1. Molecular structures of compounds 3q (left) and 4f (right). | |
{kind=link}
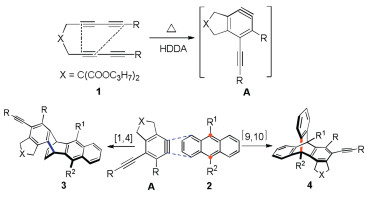
Scheme 2 indicates the sequence of steps involved in the synthesis of rigid, dihydroethenotetracene butterfly scaffolds by a cascade HDDA reaction [13] between the aryne precursors and anthracene derivatives. Tetrayne substrate 1 undergoes thermal HDDA cycloisomerization [14] to form aryne intermediate A. Anthracene reagent 2 then reacts with the aryne intermediate to form a rigid bridged compound by a formal [4 + 2] cycloaddition [15]. Stable target compounds can be produced by the cycloisomerization of tetralkyne into benzene-acetylene intermediates followed by cycloaddition. These species can easily react with anthracenes to afford the final product 3 or 4. When the substituent R2 on anthracene has small steric resistance, the addition reaction prefers to react at 1, 4-positions to generate product 4. On the contrary, the addition reaction occurs on the 9, 10-positions to obtain product 3 when substituent R2 has large steric resistance.

|
Download:
|
| Scheme 2. Tentative mechanism of the HDDA and [4 + 2] cycloaddition. | |
{kind=link}
In conclusion, we developed a new D–A reaction between tetraynes and anthracene derivatives with unusual regio- and stereo-selectivity. The typical products of the D–A reaction of anthracenes are mixtures of diastereomers. This efficient method enables the formation of adducts of anthracene derivatives with a wide substrate range, high regio- and stereo-selectivity, and satisfactory yields. At the same time, we have also investigated the influence of the electric effect and steric hindrance effect of the substituents to the yields and regioselectivity. This synthesis strategy is contrary to the traditional idea of gradual substitution from the starting material but exhibits improved efficiency for preparing polysubstituted bridge ring systems with complex stereochemical environments.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsWe greatly acknowledge the National Natural Science Foundation of China (Nos. 21572002, 21272005), The Research Culture Funds of Anhui Normal University, and Department of Human Resources of Anhui Province for financial support.
Appendix A. Supplementary dataSupplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.cclet.2019.10.003.
| [1] |
(a) A. Bhunia, S.R. Yetra, A.T. Biju, Chem. Soc. Rev. 41 (2012) 3140-3152; (b) D. Rodriguez-Lojo, D. Perez, D. Pena, E. Guitian, Chem. Commun. (Camb.) 49 (2013) 6274-6276; (c) Z. Wu, Z. Zhen, J.H. Jiang, et al., J. Am. Chem. Soc. 131 (2009) 12325-12332; (d) K. Zheng, X.H. Liu, X.M. Feng, Chem. Rev. 118 (2018) 7586-7656; (e) V. Vij, V. Bhalla, M. Kumar, Chem. Rev. 116 (2016) 9565-9627. |
| [2] |
(a) N. Zydziak, B. Yameen, C. Barner-Kowollik, Polym. Chem. 4 (2013) 4072-4086; (b) D. Wu, H.J. Ge, S.H. Liu, et al., RSC Adv. 3 (2013) 22727-22738; (c) J. Lu, D.M. Ho, N.J. Vogelaar, et al., J. Am. Chem. Soc. 128 (2006) 17043-17050; (d) G. Li, C. Piemontesi, Q. Wang, et al., Angew. Chem. Int. Ed. 58 (2019) 2870-2874; (e) A. Bunescu, Q. Wang, J.P. Zhu, Org. Lett. 16 (2014) 1756-1759. |
| [3] |
(a) I. Pozo, A. Cobas, D. Pena, et al., Chem. Commun. (Camb.) 52 (2016) 5534-5537; (b) M. Nakano, K. Takimiya, Chem. Mater. 29 (2017) 256-264; (c) Z. Chen, X. Han, J.H. Liang, et al., Chin. Chem. Lett. 25 (2014) 2535-2539; (c) Z.J. Li, W.H. Wang, H. Jian, et al., Chin. Chem. Lett. 30 (2019) 386-388. |
| [4] |
(a) J.R. Shi, Y.Y. Li, Y. Li, Chem. Soc. Rev. 46 (2017) 1707-1719; (b) D.C. Qiu, J.R. Shi, Q.Y. Guo, et al., J. Am. Chem. Soc. 140 (2018) 13214-13218; (c) E.A. Margulies, N. Kerisit, P. Gawel, et al., J. Phys. Chem. C 121 (2017) 21262-21271; (d) D. Lehnherr, R. McDonald, R.R. Tykwinski, Org. Lett. 10 (2018) 4163-4166. |
| [5] |
(a) A. Khasanov, D. Kopchuk, I. Kovalev, et al., New J. Chem. 41 (2017) 2309-2320; (b) Q.C. Zhang, J. Lv, S.J. Li, et al., Org. Lett. 20 (2018) 2269-2272. |
| [6] |
(a) S. Katsuta, D. Miyagi, H. Yamada, et al., Org. Lett. 13 (2011) 1454-1457; (b) A. Lorbach, M. Bolte, H.W. Lerner, et al., Organometallics 29 (2010) 5762-5765. |
| [7] |
(a) Q. Chen, H. Chen, X. Meng, et al., Org. Lett. 17 (2015) 5016-5019; (b) L.E. Harrington, J.F. Britten, M.J. McGlinchey, Org. Lett. 6 (2003) 787-790; (c) J.W. Wang, L.Y. Zhou, H. Sun, et al., Chem. Mater. 30 (2018) 5544-5549. |
| [8] |
(a) M. Randic, Chem. Rev. 103 (2003) 3449-3605; (b) M.E. Hayes, H. Shinokubo, R.L. Danheiser, Org. Lett. 7 (2005) 3917-3920. |
| [9] |
(a) D. Niu, P.H. Willoughby, B.P. Woods, et al., Nature 501 (2013) 531-534; (b) X. Xiao, T.R. Hoye, J. Am. Chem. Soc. 141 (2019) 9813-9818; (c) H. Shen, X. Xiao, T.R. Hoye, Org. Lett. 21 (2019) 1672-1675; (d) R. Karmakar, A. Le, P.P. Xie, et al., Org. Lett. 20 (2018) 4168-4172; (e) S. Gupta, P.P. Xie, Y.Z. Xia, et al., Org. Lett. 19 (2017) 5162-5165; (f) V.R. Sabbasani, H.J. Lee, P.P. Xie, et al., Chem. -Eur. J. 23 (2017) 8161-8165. |
| [10] |
X.Z. Meng, S. Lv, D. Cheng, et al., Chem. -Eur. J. 23 (2017) 6264-6271. DOI:10.1002/chem.201701009 |
| [11] |
(a) T. Kitamura, K. Gondo, J. Oyamada, J. Am. Chem. Soc.139 (2017) 8416-8419; (b) B.H. Liu, C.Y. Mao, Q. Hu, et al., Org. Chem. Front. 6 (2019) 2788-2791; (c) B.H. Liu, Q. Hu, Y.S. Wen, et al., Front. Chem. 7 (2019) 374-380. |
| [12] |
(a) R. Trevisan, C. Voy, S.X. Chen, et al., Environ. Sci. Technol. 14 (2019) 8405-8415; (b) Y.M. Hu, J. Ma, L.D. Li, et al., Chem. Commun. (Camb.) 53 (2017) 1542-1545; (c) Y.W. Wang, Y.X. Hua, H.H. Wu, et al., Chin. Chem. Lett. 28 (2017) 1994-1996; (d) A.M. Luo, Y.L. Shao, K.J. Zhang, et al., Chin. Chem. Lett. 28 (2017) 2009-2013. |
| [13] |
(a) S.E. Suh, S.M. Chen, K.N. Houk, et al., Chem. Sci. 9 (2018) 7688-7693; (b) J.S. Barber, M.M. Yamano, M. Ramirez, et al., Nat. Chem. Biol. 10 (2018) 953-960. |
| [14] |
(a) B.A. Vara, J.N. Johnston, J. Am. Chem. Soc. 138 (2016) 13794-13797; (b) W.C. Wertjes, M. Okumura, D. Sarlah, J. Am. Chem. Soc.141 (2019) 163-167. |
| [15] |
(a) X.Y. Chen, J.T. Merrett, P.W.H. Chan, Org. Lett. 20 (2018) 1542-1545; (b) Z. Huang, W.X. Zhang, Z.F. Xi, Org. Lett. 20 (2018) 485-488. |