 2020, Vol. 31
2020, Vol. 31 


b Division of Antitumor Pharmacology, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
c Institute for Translational Medicine, College of Medicine, Qingdao University, Qingdao 266021, China;
d Department of Biochemistry and Biophysics, Institute of Systems Biomedicine, School of Basic Medical Sciences, Peking University Health Science Center, Beijing 100191, China
Epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) have brought great benefits in the clinic for the treatment of non-small cell lung cancer (NSCLC) patients harboring EGFR-mutation. To date, three generations of EGFR inhibitors [1, 2], e.g., gefitinib, erlotinib, afatinib, dacomitinib and osimertinib (AZD9291), have been approved by Food and Drug Administration (FDA). Particularly, the third generation EGFR Thr790 to Met790 mutant (T790 M) inhibitor osimertinib, covalently binding to Cys797, demonstrated approximately 75% overall response rate (ORR) in EGFRT790M mutation-positive NSCLC patients and was approved as the first-line treatment for metastatic NSCLC patients in 2018 [3-5]. However, EGFR tertiary Cys797 to Ser797 (C797S) point mutation arises rapidly after treatment of osimertinib in clinic, due to the fact that the less reactive serine797 fails to form a covalent bond with osimertinib [6, 7]. EGFRC797S mutation has become the most common acquired resistance to the third-generation EGFR-TKIs [8]. Thus, it is highly urgent to develop the fourth-generation EGFR inhibitors to overcome EGFRC797S mutation [9].
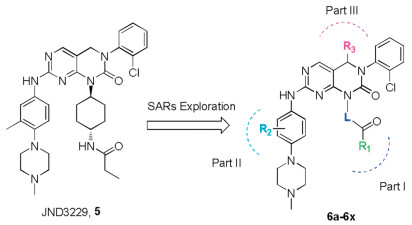
Several fourth-generation inhibitors of EGFR with different binding types have been reported [1, 10], including the allosteric inhibitors EAI045 (1a) and JBJ-04-125-02 (1b) [11], and the ATPcompetitive inhibitors 2-4 [12-15] (Fig. 1). Among them, the (2-hydroxy)pheyl-4-substitututed quinazoline 2 and tri-substituted imidazole 3 displayed strong potency against EGFRC797S kinase in biochemical assay, but neither cell-based activity nor in vivo efficacy of these molecules was disclosed. EAI045 (1a) and brigatinib (4) were both required to combine with EGFR antibody cetuximab or pantitumumab to demonstrate in vivo therapeutic efficacy [11, 15]. Most recently, we have identified a pyrimidopyrimidinone-based derivative JND3229 (5) as a new reversible EGFRC797S mutant inhibitor demonstrating both in vitro and in vivo mono-drug efficacy against the proliferation of EGFRC797S mutated cancer cells [16]. Structure analysis indicated that JND3229 bound to the ATP binding site of EGFRC797S with a reversible "U-shaped" configuration. The 2-oxo-3, 4-dihydropyrimido[4, 5-d]pyrimidine privileged core formed a bidentate hydrogen bond interactions with Met793, the 2-chlorophenyl group was directed toward the hydrophobic back pocket, and the carbonyl in pyrido[4, 5-d] pyrimidine formed a hydrogen bond with Lys745 mediated by a water molecule. Herein, we would like to describe extensive structure-activity relationships (SARs) of this 2-oxo-3, 4-dihydropyrimido[4, 5-d]pyrimidine privileged scaffold based compounds (Fig. 2).

|
Download:
|
| Fig. 1. The reported fourth generation EGFR inhibitors overcoming EGFRC797S mutation. | |

|
Download:
|
| Fig. 2. The designed 2-oxo-3, 4-dihydropyrimido[4, 5-d] pyrimidines privileged scaffold based on JND3229. | |
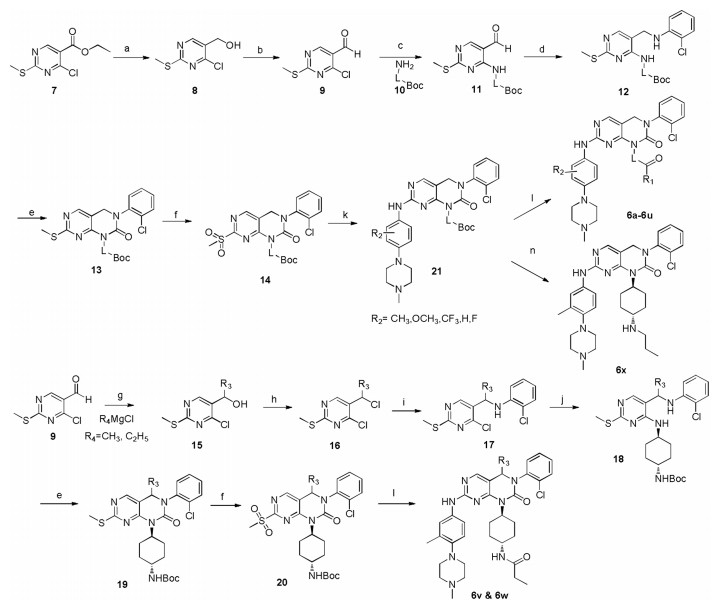
The synthetic routes of the designed 2-oxo-3, 4-dihydropyrimido [4, 5-d]pyrimidinyl analogues were outlined in Scheme 1. Briefly, reduction of 4-chloro-2-(methylthio)-pyrimidine-5-carboxylate (7) with diisobutyl aluminium hydride (DIBAL) gave (4-chloro-2-(methyl thio)pyrimidin-5-yl)methanol, which were subsequently oxidized by MnO2 to yield aldehyde 9. For compounds 6a-6u and 6x, 9 then reacted with BOC-protected aliphatic amines, followed by Borch reductive amination, cyclization and oxidation to yield key pyrimidopyrimidinone 14. For compounds 6v and 6w, 9 went through Grignard reaction to give alcohol 15, which followed by chlorination, nucleophilic substitution, cyclization and oxidation to yield key pyrimidopyrimidinone 20. The intermediates 14 and 20 were respectively subjected to nucleophilic, deprotection and acylation reaction to give the title compounds 6a-6w.

|
Download:
|
| Scheme 1. General procedures for synthesis of compounds 6a-6u, 6x, 6v and 6w. Reagents and conditions: (a) DIBAL, THF, 78 ℃, 3–4 h, 88%; (b) MnO2, DCM, r.t., overnight, 86%; (c) CH3CN, r.t., overnight, 70%–82%. (d) ⅰ) 2-chloroaniline, AcOH, PhCH3, 110 ℃. ⅱ) NaBH4, THF, 70 ℃, 50%–60%; (e) Triphosgene, DIPEA, DCM, 0 ℃, 70%–80%; (f) m-CPBA, DCM, r.t., 70%–80%; (g) R3MgCl, THF, -40 ℃, 80%–90%; (h) DIPEA, POCl3, 0 ℃, 2 h, 70%–80%. (i) 2-chloroaniline, Acetonitrile, r.t., overnight, 80%–85%; (j) 10, CH3CN, r.t., overnight, 80%–90%; (k) 3-methyl-4-(4-methylpiperazin-1-yl)aniline, TFA, 2-butyl alcohol, 110℃, overnight, 40%–50%; (l) ⅰ) TFA, DCM, r.t.; ⅱ) propionic acid, HATU, DIPEA, DCM, overnight, 30%–50%. (n) ⅰ) TFA, DCM, r.t.; ⅱ) iodine propane, EtOH, DCM, 80℃, 20 h; 28%–35%. | |
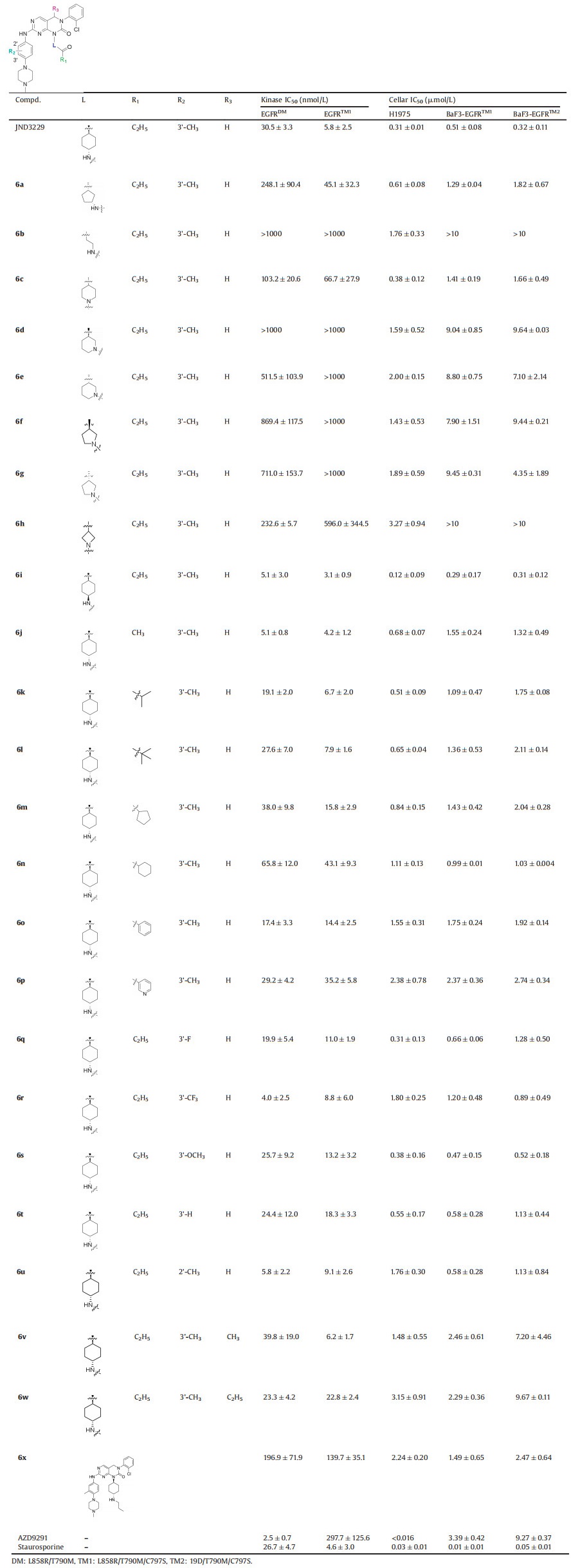
The kinase inhibitory activities of the title compounds were evaluated by the enzyme-linked-immunosorbent assay (ELISA) assay (Table 1). Osimertinib, staurosporine and JND3229 were used as positive controls to validate the screening conditions. As shown in Table 1, osimertinib potently inhibits EGFRL858R/T790M, while is less potent against EGFRL858R/T790M/C797S. However, staurosporine, a broad spectrum of multi-kinase inhibitor, suppresses the enzymatic activity of the EGFRL858R/T790M and EGFRL858R/T790M/C797S mutants with low nanomolar IC50 values.
|
|
Table 1 In vitro EGFR inhibition and anti-proliferation data of compounds 6a-6x. |
{kind=link}
{kind=link}
{kind=link}
We firstly investigated the SARs on the linker L (Fig. 2) and the results were summarized in Table 1. Change of trans-cyclohexanamine in JND3229 with a cis-cyclopyrrilidinamine (6a) and linear ethylamine (6b) led to 14-fold and complete potency loss against EGFRL858R/T790M/C797S. When exocyclic N atom in the linker was incorporated into the cycloalkyl group, the resulting compounds (6c–6h) showed greatly decreased potency on EGFRL858R/T790M/C797S. Compound 6i with cis-cyclohexanamine instead of the trans-cyclohexanamine exhibited an IC50 value of 3.1 nmol/L against EGFRL858R/T790M/C797S, which is comparable to that of JND3229. Moreover, this compound is 7-fold more potent than JND3229 with the inhibitory activity of EGFRL858R/T790M double mutant. These results indicated that the cyclohexanamine group in the linker fraction was preferable for the high inhibitory activity.
After determined that the cyclohexanamine was the optimal substituent for the L part, we then turned to optimize the R1 substitution (Table 1). It was revealed that the ethyl in JND3229 could be replaced with a variety of relative small substituents (e.g., methyl (6j), isopropyl (6k), tert-butyl (6l)), without obviously affecting the EGFR inhibitory potency. However, when the ethyl was replaced with larger hydrophobic groups, including cyclopentyl (6m), or cyclohexyl (6n), and phenyl (6o), the resulting molecules displayed about 3- to 7-folds potency loss. For instance, 6o with the phenyl substituent exhibited an IC50 value of 14.4 nmol/L against EGFRL858R/T790M/C797S. Interestingly, introducing an N atom into this phenyl group in 6o further reduced the compound's activity (6p, IC50 = 35.2 nmol/L). It was worthy to note that when the propionyl moiety in JND3229 was replaced with a propyl group, the resulting compound (6x) dramatically decreased the kinase inhibitory potency with an IC50 value of 139.7 nmol/L against EGFRL858R/T790M/C797S. The investigation suggested that a critical interactionmight exist between the carbonyl group and the protein.
The X-ray structure analysis of EGFRT790M/C797S and JND3229 complex revealed that the methyl substituted phenylpiperazine moiety of JND3229 was exposed to a solvent accessible surface [16]. We then explored the preliminary SARs of 3'-methyl substituent. When the 3'-methyl group in JND3229 was replaced with 3'-CF3 (6r) or changed to 2'-position (6u), the resulting compounds exhibited comparable activities, while replacement of 3'-methyl group with the other substituents, e.g., 3'-F (6r), 3OCH3 (6s), or removal of it (6t), yielded compounds with potency loss ranging from 2- to 3-folds.
At last, we investigated the potential steric impact of R3 position by substitution with methyl (6v) and ethyl (6w). The compound 6v exhibited the equal activity compared to the parental compound JND3229, while the ethyl substituted compound 6w displayed about 4-folds potency loss, suggesting that the bulky moieties were not tolerated at this site.
In order to get insight into the binding mode of cys-cyclohexanamine with EGFRT790M/C797S, a co-crystal structure of 6i complexed with EGFRT790M/C797S was determined (Fig. 3A and Table S1 in Supporting information). It was indicated that 6i bound to EGFRT790M/C797S with a reversible "U-shaped" configuration with the "hinge" residue Met793. The 2-chlorophenyl group was directed toward the hydrophobic back pocket composed by Lys745, Glu762, Leu788, Met766 and Met790. Different conformations of cyclohexylamine caused the different binding patterns to protein. The carbonyl of amide in 6i formed a hydrogen bond with Ser720 mediated by a water molecule, while the NH of amide in JND3229 formed a hydrogen bond with Leu718 mediated by another water molecule.

|
Download:
|
| Fig. 3. (A) The X-ray crystal structure of 6i with EGFRT790M/C797S (PDB ID: 6JRX). (B) Overlay the 6i and JND3229 in EGFRT790M/C797S (PDB ID: 5ZTO). The EGFR kinase is shown in green and blue stick and ribbon representation. 6i and JND3229 are shown in purple and yellow stick. Hydrogen bonds are indicated by yellow and black hatched lines to key amino acids. Water is represented as red dots. | |
{kind=link}
The antiproliferative activities of compounds 6a-6x were also investigated against H1975 (EGFRL858R/T790M) and BaF3 cells stably transfected with EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S mutants (Table 1). It was showed that 6a-6x exhibited moderate antiproliferative activities against H1975 and BaF3 cells harboring EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S mutants, with IC50 values of 0.12–3.27 μmol/L, 0.29->10μmol/L and 0.31->10 μmol/L, respectively. Compound 6i displayed the most potent antiproliferative activities against BaF3-EGFRL858R/T790M/C797S and BaF3-EGFR19D/T790M/C797S with IC50 values of 0.29 μmol/L and 0.31 μmol/L, respectively, which is corresponding to that of biochemical kinase assay.
Further, the kinase inhibition of the most potent compounds 6i and 6s (both showed sub-micromolar activities against BaF3-EGFRL858R/T790M/C797S and BaF3-EGFR19D/T790M/C797S) was further validated by investigating its suppressive functions on the activation of EGFR in BaF3-EGFRL858R/T790M/C797S and BaF3EGFR19D/T790M/C797S mutants. As shown in Fig. 4, compounds 6i and 6s caused a dose-dependent suppression of the phosphorylation of EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S starting from the concentration of 100 nmol/L.

|
Download:
|
| Fig. 4. Compounds 6i and 6s potently suppressed the phosphorylation of EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S in BaF3 cells. Cells were treated with or without compounds 6i and 6s for 4 h at indicated concentration, respectively. Cells were then stimulated by 50 ng/mL EGF for 10 min and harvested for western blot analysis. | |
{kind=link}
In summary, an extensive SAR investigation was conducted based on our recently disclosed 2-oxo-3, 4-dihydropyrimido[4, 5-d] pyrimidine privileged scaffold-based EGFRC797S inhibitor JND3229. One of the most potent compound 6i potently suppressed EGFRL858R/T790M/C797S kinase and inhibited the proliferation of BaF3 cells harboring EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S mutants with IC50 values of 3.1 nmol/L, 290 nmol/L and 316 nmol/L, respectively. Compound 6i dose-dependently induced suppression of the phosphorylation of EGFRL858R/T790M/C797S and EGFR19D/T790M/C797S in BaF3 cells starting from the concentration of 100 nmol/L. Compound 6i may serve as a new generation of EGFR inhibitors for anticancer drug discovery.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThe authors appreciate the financial support from the National Natural Science Foundation of China (Nos. 81922062, 81874285 and 81673285), Guangdong International Science and Technology Cooperation Project (No. 2018A050506043), Guangzhou City Key Laboratory of Precision Chemical Drug Development (No. 201805010007) and Institutes for Drug Discovery and Development of Chinese Academy of Science (No. CASIMM0120185006).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.044.
| [1] |
X. Lu, L. Yu, Z. Zhang, et al., Med. Res. Rev. 38 (2018) 1550-1581. DOI:10.1002/med.21488 |
| [2] |
Z. Song, Y. Ge, C. Wang, et al., J. Med. Chem. 59 (2016) 6580-6594. DOI:10.1021/acs.jmedchem.5b00840 |
| [3] |
D.A. Cross, S.E. Ashton, S. Ghiorghiu, et al., Cancer Discov. 4 (2014) 1046-1061. DOI:10.1158/2159-8290.CD-14-0337 |
| [4] |
S.L. Greig, Drugs 76 (2016) 263-273. DOI:10.1007/s40265-015-0533-4 |
| [5] |
J.C. Soria, Y. Ohe, J. Vansteenkiste, et al., N. Engl. J. Med. 378 (2018) 113-125. DOI:10.1056/NEJMoa1713137 |
| [6] |
Z. Piotrowska, M.J. Niederst, C.A. Karlovich, Cancer Discov. 5 (2015) 713-722. DOI:10.1158/2159-8290.CD-15-0399 |
| [7] |
K.S. Thress, C.P. Paweletz, E. Felip, et al., Nat. Med. 21 (2015) 560-562. DOI:10.1038/nm.3854 |
| [8] |
M.J. Niederst, H. Hu, H.E. Mulvey, et al., Clin. Cancer Res. 21 (2015) 3924-3933. DOI:10.1158/1078-0432.CCR-15-0560 |
| [9] |
T. Grabe, J. Lategahn, D. Rauh, et al., ACS Med. Chem. Lett. 9 (2018) 779-782. DOI:10.1021/acsmedchemlett.8b00314 |
| [10] |
L. Chen, W. Fu, L. Zheng, et al., J. Med. Chem. 61 (2018) 4290-4300. DOI:10.1021/acs.jmedchem.7b01310 |
| [11] |
(a) Y. Jia, C.H. Yun, E. Park, et al., Nature 534(2016) 129-132; (b) C. To, J. Jang, T. Chen, et al., Cancer Discov. 9(2019) 926-943. |
| [12] |
H. Park, H.Y. Jung, S. Mah, S. Hong, Angew Chem. Int. Ed. Eng. 56 (2017) 7634-7638. DOI:10.1002/anie.201703389 |
| [13] |
M. Gunther, M. Juchum, G. Kelter, et al., Angew Chem. Int. Ed. Eng. 55 (2016) 10890-10894. DOI:10.1002/anie.201603736 |
| [14] |
M. Juchum, M. Gunther, E. Doring, et al., J. Med. Chem. 60 (2017) 4636-4656. DOI:10.1021/acs.jmedchem.7b00178 |
| [15] |
K. Uchibori, N. Inase, M. Araki, Nat. Commun. 8 (2017) 14768.
|
| [16] |
X. Lu, T. Zhang, S. Zhu, ACS Med. Chem. Lett. 9 (2018) 1123-1127. DOI:10.1021/acsmedchemlett.8b00373 |