 2020, Vol. 31
2020, Vol. 31 


b School of Life Sciences, University of Science and Technology of China, Hefei 230027, China;
c Jiangsu Key Laboratory of Neuropsychiatric Diseases and College of Pharmaceutical Sciences, Soochow University, Suzhou 215123, China
Histones including H2A, H2B, H3, and H4 are pivotal components of chromatin [1]. Around the four core histones, varieties of PTMs (post-transcriptional modifications) have been identified, including methylation, acetylation, phosphorylation and ubiquitination [2]. Histone PTMs contribute to regulation of genes transcription either by directly altering the chromatin structure or by providing a binding platform for chromatin-related factors [3]. Moreover, the crosstalk and combinatorial readout of histone PTMs further strengthen the accuracy and complexity of the regulation of chromatin structure and gene transcription [4]. For example, the H3K79 targeting activity of methyl transferase hDot1L is directly stimulated by H2B Lys120 ubiquitination (H2BK120Ub) [5]. Correspondingly, as a synergetic pair of PTMs, K9Me3 and K18Ac of histone H3 (H3-K9Me3-K18Ac) are speculat-ed to collectively recruit the PTMs-specific protein TRIM33 [6] in the nodal signalling pathway [7].
The nodal signaling pathway is critical for the differentiation of embryonic stem cells (ESCs) [8]. At the early stage of ESCs differentiation [7a], nodal proteins binding to activin and activin-like proteins triggers the phosphorylation of Smad2 protein. Subsequently the phosphorylation signal promotes the formation of Smad2/3 and Smad2/3/4 complexes, which bind to activin response elements (AREs) in the proximal promoters of Gsc and Mixl1 [9], thus activating the poised state of genes from HP1g suppression [10], and the gene transcription would significantly affect gastrulation, axial mesendoderm morphogenesis, and endoderm formation. As an indispensable component to activate regulator genes, H3-K9Me3-K18Ac recruits Smad2/3 to chromatin by the complexes of TRIM33 and Smad2/3. The dual marks are supposed to be recognized by the plant homeodomain (PHD) and Bromo domain of TRIM33 respectively [7a]. This recognition mechanism recently has been revealed by the crystal structure of PHD-Bromo cassette (truncated TRIM33, sequence: 882-1087) bound to bivalently modified H3 N-terminal peptide [7a].
However, the interaction details between TRIM33 and the bivalently modified H3 at the nucleosome level and whether the H3 tail could substitute the nucleosome to elucidate the interaction still need further exploration. For these purposes, the pivotal procedure is to obtain the H3 bearing bivalent modifica-tions of K9Me3 and K18Ac, which cannot be isolated from natural sources or prepared through enzymatic methods.
Here we reported an improved three-segment ligation strategy for total chemical synthesis of bivalently modified histone H3. CD (circular dichroism) spectroscopy of H3-K9Me3-K18Ac dem-onstrated structural alteration by the bivalent modification. Furthermore, we successfully reconstituted it into NCPs with good homogeneity, which may be applicable for biochemical and biophysical studies in the future.
Recently, great efforts have been made for semi-synthesis or total chemical synthesis of modified histones [11]. Especially, histone total chemical synthesis gained much attention because of the less limiting in the synthesis of modified histone, regardless of PTMs types, positions and numbers [12]. Ottesen's group [13] and Liu's group [14] previously illustrated the total chemical synthesis of histone H3 variant containing one Cysteine at site 110 by twice ligations of three segments. However, the middle segment was not liable to accomplish with acceptable yield. Recently Ashraf's group [15] also synthesized varieties of modified H3 analogues (Homo sapiens). To avoid the low yield of segment synthesis, they divided the sequence into four segments, and combined them by three steps of native chemical ligation (NCL) and retained the auxiliary thiol group after NCL, which would consequently complicate the process and influence the overall yield [16]. For preparing H3-K9Me3-K18Ac with acceptable yield in a convenient route, we decided to adopt an alternative strategy with an improved three-segment ligation, which is specifically illustrated in Scheme 1.

|
Download:
|
| Scheme 1. The scheme for the total chemical synthesis of histone H3-K9Me3-K18Ac. MPAA: 4-mercaptophenylacetic acid; VA-044: 2, 2'-azobis-[2-(2-imidazolin-2-yl) propane] dihydrochloride; TCEP: Tris(2-carboxyethyl)phosphine. PDB: 2hue. | |
{kind=link}
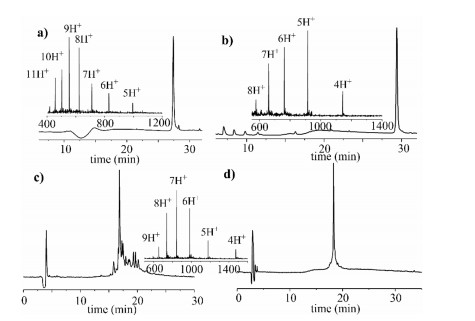
The 135 amino acids (AAs) of H3 were divided into three segments, including H3-K9Me3-K18Ac(1-46)-NHNH2 (1), Cys-H3 (48-95)-NHNH2 (2), and Cys-H3(97-135)-OH (3), which would be sequentially ligated by hydrazide-based NCL [17]. All segments were anticipated to be synthesized by (9H-fluoren-9-ylmethoxy) carbonyl solid-phase peptide synthesis (Fmoc-SPPS). After SPPS completed, the peptide segments were cleaved from loaded resin by TFA cocktail (5% thioanisole, 5% H2O, 3% ethanedithiol, 87% TFA) in 3 h. Peptide segments 1 and 3 were comparatively easily synthesized that were sequentially purified by semi-preparative RP-HPLC (reverse phase high-performance liquid chromatogra-phy) and verified by ESI-MS (electrospray ionization mass spectrometry) (Figs. 1a and b). Finally, we acquired segment 1 and segment 3 with overall isolated yield 30% and 35%.

|
Download:
|
| Fig. 1. Synthesis of peptide segments of H3-K9Me3-K18Ac. (a) Analytical HPLC trace (214 nm) and ESI-MS characterization (Obs.: 4969.6 Da, Calcd.: 4970.6 Da, average isotopes) of purified segment 1. (b) Analytical HPLC trace (214 nm) and ESI-MS characterization (Obs.: 4572.5 Da, Calcd.: 4572.3 Da average isotopes) of purified segment 3. (c) Analytical HPLC trace (214 nm) of crude peptide segment 2 after incorporating Fmoc-Ser [Psi(Me, Me)pro]-OH. (d) Purified segment 2 HPLC trace (214 nm) and ESI-MS characterization (Obs.: 5892.9 Da, Calcd.: 5890.9 Da, average isotopes). | |
{kind=link}
During our synthesizing Cys-H3(48-95)-NHNH2 (2) according to the conventional one by one amino acid coupling strategy, we encountered an obstacle that several amino acids were coupled with low efficiency, which severely influenced the overall isolated yield. Double coupling of all amino acids and prolonging the reaction time simultaneously were also attempted, but the situation was not improved.
Through analytical RP-HPLC trace (214 nm) analysis, we found that several peaks that referred to the broken peptide fragments (Fig. S2 in Supporting information). Considering the phenomenon started to emerge after the Ser-Ser site, we intended to incorporate Fmoc-Ser(tBu)-Ser (Psi(Me, Me)pro)-OH building block [18] (Scheme 1) into peptide synthesis to perturb the secondary structure, and the remaining residues were double coupled. The Met90 was also mutated to norleucine (Nle) to prevent oxidation during Fmoc-SPPS [15]. To our satisfaction, segment 2 was successfully synthesized (Fig. 1c) and purified (Fig. 1d) with preferable isolated yield (30%) compared with before (5%, even less). Therefore, this pseudo dipeptide strategy settled the dilemma in Cys-H3(47-95)-NHNH2 (2) synthesis, and may be extensible to conquer the drawback in previous three-segment division strategy [13, 14].
With all three segments in hand, we firstly conducted the hydrazide-based NCL between segment 1 and segment 2 (Fig. 2a). Segment 1 (1.3 mmo/L, 1.3 equiv.) was firstly dissolved in ligation buffer (6 mol/L Gn HCl, 200 mmol/L NaH2PO4, pH 2.8), and treated with 7 equiv. NaNO2 to oxide the hydrazide at -10 ℃. 30 min later, MPAA (40 equiv.) was added to convert the C-terminal of segment 1 into thioester following adjusting pH to 5.0. When the thioester transition completed (5 min), segment 2 (1 mmol/L) was added to proceed ligation step, which ultimately furnished segment 4 with 40% isolated yield in 48 h. The subsequent procedure was the desulfurization of segment 4 (1 mmol/L) that converting the mutated Cys47 back to the Ala residue. Under the condition of desulfurization [19] (250 mmol/L TCEP, 10% (v/v) tBu-SH, 50 mmol/L VA-044, pH 6.8, 37 ℃), the total conversion of segment 4 into segment 5 was completed in 4 h with 75% isolated yield. The last ligation procedure between segment 3 and segment 5 also exactly obeyed the process of hydrazide-based NCL mentioned above. The reaction proceeded promptly within 4 h to furnish final product. The final status of each process before purification was presented in Fig. 2a. The purified H3-K9Me3-K18Ac was charac-terized by analytical RP-HPLC and high-resolution ESI-MS (Fig. 2b). Eventually, H3-K9Me3-K18Ac (6) was obtained with 12% overall isolated yield on the tens of milligrams scale.

|
Download:
|
| Fig. 2. (a) Analytical HPLC-traces (214 nm) for the ligations and desulfurization reaction. 1' was the MPAA thioester of 1. # referred to MPAA. * referred to hydrolysis product. Segment 1 and 2 ligation and desulfurization gradient: 20%-60% acetonitrile (0.1% TFA) in water (0.1% TFA) over 35 min (20% for 5 min, then 20%-60% for 30 min). The ligation of segment 3 and 5 was proceeded at linear gradient: 20%-80% acetonitrile (0.1% TFA) in water (0.1% TFA) over 32 min (20% for 2 min, then 20%-80% for 30 min). (b) Analytical HPLC trace (214 nm) and ESI-MS characteriza-tion of purified H3-K9Me3-K18Ac. | |
{kind=link}
To analyse the secondary structure, the synthetic H3-K9Me3-K18Ac was examined by CD spectroscopy (Fig. 3a), which exhibited prominent negative peak at 200 nm. Compared to the negative peak of recombinant H3 at 206 nm, this shorter-wavelength shift may be attributed to the influence of tri-methylation and acylation that induce structural alterations of H3 protein. This phenomenon resulted from PTMs was also discovered by Namatame's group, while analysing the lysine-4 methylated histone H3 [20].

|
Download:
|
| Fig. 3. (a) CD spectroscopy of the synthetic H3-K9Me3-K18Ac and recombinant H3 (25 ℃, 0.2 mg/mL). (b) FPLC-trace of purified bivalently modified nucleosomes by Superose 6. (c) Gel mobility shift assay of reconstituted NCPs containing H3-K9Me3-K18Ac. | |
{kind=link}
To test the biochemical properties of the synthetic histone H3, we sequentially reconstituted it into octamers and nucleosomes together with recombinant histone H2A, H2B and H4. Histones were separately dissolved in unfolding buffer (6 mol/L Gn HCl, 200 mmol/L Tris HCl, 5 mmol/L DTT, pH 7.5) and subsequently mixed and dialyzed into refolding buffer (2 mol/L NaCl, 10 mmol/L Tris HCl 1 mmol/L EDTA, 5 mmol/L 2-hydroxy-1-ethanethiol, pH 7.5) at 4 ℃. Resulted octamers were purified by size-exclusive chromatography (Fig. S5 in Supporting information). Purified octamers were concentrated and further reconstituted into NCPs with 147 base pair of 601 DNA chains by gradient salt dialysis [21]. The results showed that the assembled NCPs were highly homogenous, which indicated that the dual marks did not interfere with NCPs reconstitution in vitro (Fig. 3b). In gel mobility shift assay (Fig. 3c), while free DNA located under 200 bp, the band of reconstituted NCPs shifted over 300 bp, indicating the binding of octamers and 601 DNA. Thus, it confirmed the success of nucleosome reconstitution and exhibited good homogeneity.
In summary, based on the efficient synthesis of H3 segment with pseudo dipeptide incorporation, we have developed a practical method for three-segment-directed total chemical synthesis of H3-K9Me3-K18Ac. CD spectroscopy of H3-K9Me3-K18Ac demonstrated the alterations of secondary structure by these bivalent modifications, and finally we successfully recon-stituted it into NCPs with good homogeneity. This synthetic bivalently modified nucleosomes could be further utilized in research of the interaction with TRIM33. We also envisioned that this new strategy would serve as a valuable tool in the synthesis of other multi-valently modified histone H3 proteins.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21708036, 31470740, U1732161) and Anhui Provincial Natural Science Foundation (No. 1808085QC63)
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.038.
| [1] |
K. Luger, A.W. Mader, R.K. Richmond, et al., Nature 389 (1997) 251-260. DOI:10.1038/38444 |
| [2] |
(a) H. Huang, B.R. Sabari, B.A. Garcia, et al., Cell 159(2014) 458-458; (b) H. Huang, S. Lin, B.A. Garcia, et al., Chem. Rev. 115(2015) 2376-2418. |
| [3] |
M. Tan, H. Luo, S. Lee, et al., Cell 146 (2011) 1016-1028. DOI:10.1016/j.cell.2011.08.008 |
| [4] |
L.M. Soares, S. Buratowski, Mol. Cell 49 (2013) 1019-1020. DOI:10.1016/j.molcel.2013.03.012 |
| [5] |
(a) H.H. Ng, R.M. Xu, Y. Zhang, et al., J. Biol. Chem. 277(2002) 34655-34657; (b) S.D. Briggs, T. Xiao, Z.W. Sun, et al., Nature 418(2002) 498; (c) R.K. McGinty, J. Kin, C. Chatterjee, et al., Nature 453(2008) 812-816; (d) T. Yao, W. Jing, Z. Hu, et al., Cell Res. 29(2019) 330-333; (e) E.J. Worden, N.A. Hoffmann, C.W. Hicks, et al., Cell 176(2019) 1490-1501; (f) C.J. Anderson, M.R. Baird, A. Hsu, et al., Cell Rep. 26(2019) 1681-1690. |
| [6] |
S. Dupont, L. Zacchigna, M. Cordenonsi, et al., Cell 121 (2005) 87-99. DOI:10.1016/j.cell.2005.01.033 |
| [7] |
(a) Q. Xi, Z. Wang, A.I. Zaromytidou, et al., Cell 147(2011) 1511-1524; (b) L. Morsut, K.P. Yan, E. Enzo, et al., Development 137(2010) 2571-2578. |
| [8] |
J. Brennan, C.C. Lu, D.P. Norris, et al., Nature 411 (2001) 965-969. DOI:10.1038/35082103 |
| [9] |
W. He, D.C. Dorn, H. Erdjument-Bromage, et al., Cell 125 (2006) 929-941. DOI:10.1016/j.cell.2006.03.045 |
| [10] |
L. Izzi, C. Silvestri, I. von Both, et al., EMBO J. 26 (2007) 3132-3143. DOI:10.1038/sj.emboj.7601753 |
| [11] |
(a) M. Morgan, M. Jbara, A. Brik, et al., Methods Enzymol. 618(2019) 1-27; (b) M. Jbara, H. Sun, G. Kamnesky, et al., Curr. Opin. Chem. Biol. 45(2018) 18-26; (c) S.K. Maity, M. Jbara, G. Mann, et al., Nat. Protoc. 12(2017) 2293-2322; (d) F. Wojcik, G.P. Dann, L.Y. Beh, et al., Nat. Commun. 9(2018) 1394; (e) L.A. Farrelly, R.E. Thompson, S. Zhao, et al., Nature 567(2019) 535-539. |
| [12] |
(a) M. Jbara, S.K. Maity, M. Morgan, et al., Angew. Chem. Int. Ed. 55(2016) 4972-4976; (b) M. Seenaiah, M. Jbara, S.M. Mali, et al., Angew. Chem. Int. Ed. 54(2015) 12374-12378; (c) J. Li, Q. He, Y. Liu, et al., Chembiochem 18(2017) 176-180; (d) S. Bondalapati, M. Jbara, A. Brik, Nat. Chem. 8(2016) 407-418. |
| [13] |
J.C. Shimko, J.A. North, A.N. Bruns, et al., J. Mol. Biol. 408 (2011) 187-204. DOI:10.1016/j.jmb.2011.01.003 |
| [14] |
J. Li, Y. Li, Q. He, et al., Org. Biomol. Chem. 12 (2014) 5435-5441. DOI:10.1039/C4OB00715H |
| [15] |
(a) M. Jbara, N. Guttmann-Raviv, S.K. Maity, et al., Bioorg. Med. Chem. 25(2017) 4966-4970; (b) M. Jbara, S.K. Maity, A. Brik, Eur. J. Org. Chem. (2019), doi: http://dx.doi.org/10.1002/ejoc.201900257. |
| [16] |
P.E. Dawson, T.W. Muir, I. Clark-Lewis, et al., Science 266 (1994) 776-779. DOI:10.1126/science.7973629 |
| [17] |
(a) G.M. Fang, Y.M. Li, F. Shen, et al., Angew. Chem. Int. Ed. 50(2011) 7645-7649; (b) G.M. Fang, J.X. Wang, et al., Angew. Chem. Int. Ed. 51(2012) 10347-10350; (c) J.S. Zheng, S. Tang, Y.K. Qi, et al., Nat. Protoc. 8(2013) 2483-2495; (d) S. Tang, Y.Y. Si, Z.P. Wang, et al., Angew. Chem. Int. Ed. 54(2015) 5713-5717; (e) M. Pan, S. Gao, Y. Zheng, et al., J. Am. Chem. Soc. 138(2016) 7429-7435; (f) X.Q. Guo, J. Liang, Y. Li, et al., Chin. Chem. Lett. 29(2018) 1139-1142. |
| [18] |
P.W.R. Harris, R. Kowalczyk, D.L. Hay, et al., Int. J. Pept. Res. Ther. 19 (2013) 147-155. DOI:10.1007/s10989-012-9325-9 |
| [19] |
Q. Wan, S.J. Danishefsky, Angew. Chem. Int. Ed. 46 (2007) 9248-9252. DOI:10.1002/anie.200704195 |
| [20] |
Y. Izumi, K. Matsuo, H. Namatame, Chirality 30 (2018) 536-540. DOI:10.1002/chir.22849 |
| [21] |
P.T. Lowary, J. Widom, J. Mol. Biol. 276 (1998) 19-42. DOI:10.1006/jmbi.1997.1494 |