 2020, Vol. 31
2020, Vol. 31 


b Qinghai Provincial Key Laboratory of Tibetan Medicine Research and Key Laboratory of Tibetan Medicine Research, Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining 810008, China;
c Institute for Organic Chemistry, University of Duisburg-Essen, Essen 45117, Germany;
d College of Pharmacy, Nantong University, Nantong 226001, China
Peptide amphiphiles are one of the most attractive building blocks to create functional supramolecular materials for biomedical applications, e.g., for molecular imaging, disease diagnosis or drug/gene delivery, due to their inherent biocompatibility and biodegradability [1-4]. A variety of nanostructural assemblies including fibers, micelles, and ribbons can be formed by peptide amphiphiles and their biological functions have been extensively exploited in recent decades [5, 6]. In particular, a detailed understanding of well-organized α-helix or β-sheet secondary structures formed by peptide amphiphiles might provide a better insight into the mechanism of protein folding, which is often closely associated with many diseases [7, 8]. For example, amyloid fibrils formed via a misfolding in β-sheet-type conformations are related to Alzheimer's disease, Parkinson's disease, and other neurodegenerative disorders [9, 10]. Therefore, the rational design of small peptides which adopt a well-defined and precisely controlled secondary structure is highly significant.
Reversible non-covalent interactions, such as hydrogen bonding, electrostatic interactions, steric effects, and π-π stacking, play an important role for the formation of secondary structures in proteins and dynamic architectures [11-14]. Consequently, peptide conjugates which contain artificial binding motifs with an enhanced binding efficiency can promote the formation of highly ordered and stable secondary structures. For example, the guanidiniocarbonylpyrrole (GCP) group has proven to possess a high binding affinity toward carboxylates and other oxoanions in aqueous solutions [15]. The rigid and planar conformation of GCP allows for hydrogen bond assisted ion pairing with carboxylates. For this reason, GCP units have significantly stronger binding to carboxylates than simple N-terminal cations as in natural amino acids [16]. Moreover, the aromatic pyrrole unit in the GCP group can provide additional π-π stacking interactions to facilitate the self-assembly of peptide amphiphiles. Therefore, the GCP-motif is an ideal candidate for fabricating interesting supramolecular assemblies with well-defined secondary structures.
In our previous work [17], we have found that the GCPfunctionalized alanine tetramer (peptide 2, Scheme 1) adopts an α-helix conformation and further self-assembles into different superstructures depending on the overall concentrations under neutral conditions. In most cases, the length and amphiphilicity of the peptide skeleton have a remarkable effect on the secondary structure and the morphology of the further self-assembled nanostructures. Generally, peptides that are prone to form β-sheets are around 16–20 amino acids long and often feature alternating patterns of hydrophobic and polar amino acids. In contrast, α-helices are preferentially formed by relatively short amino acid sequences [18, 19]. Thus, we envisaged the regulation of the secondary structure through adjusting and controlling the length of GCP-containing peptide chains and explored their ability to self-assemble into well-defined supramolecular structures.

|
Download:
|
| Scheme 1. Schematic illustration of the formation of different secondary structures and morphologies by supramolecular self-assembly. | |
{kind=link}
Herein, we report the design and synthesis of three GCPcontaining peptide amphiphiles with different backbone length, and found that their secondary structures and morphologies can be well regulated under neutral environment. As shown in Scheme 1, peptide 1 adopted random coil conformation and further selfassembled into left-handed twisted helical fibers at high concentration. However, α-helix and β-sheet conformations were obtained for peptide 2 and peptide 3, respectively. For peptide 2, the α-helix conformation led to the formation of longer bamboolike nanostructures. But for peptide 3, an obvious aging effect could be observed with morphological transition from nanoparticles to disordered fibers after storing the freshly prepared samples for one week.
Three GCP-containing peptide amphiphiles (peptide 1-3) with different backbone length were obtained by conventional Fmoc solid-phase peptide synthesis [20]. In brief, peptide 1 was synthesized by coupling of the GCP group with an alanine dimer. Instead of an alanine dimer, the corresponding alanine tetramer or alanine hexamer were used for peptide 2 and peptide 3, to couple with the GCP group (for details, see Supporting information).
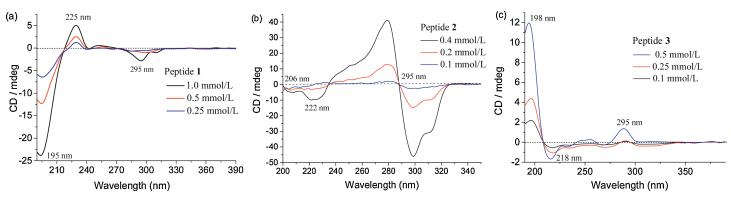
Because alanine has a strong preference to form helix-type structures in aqueous solution, but it is also a weak β-sheet former, we first investigated the conformation of these peptide amphiphiles under neutral conditions (pH 7.0) by circular dichroism (CD) spectroscopy. As shown in Fig. 1, a negative band at 195 nm and a positive band at 225 nm appeared for peptide 1. This proved the formation of random coil conformation [21], and the weak negative peak at 295 nm corresponds to the absorption maximum of the GCP unit. However, a completely different CD spectrum was observed for peptide 2, in which the GCP chromophore gave rise to a bisignate Cotton curve centered at its absorption maximum first with a negative band at 299 nm, and then with a positive band at 279 nm. Moreover, with increasing concentration of peptide 2, the CD intensities at both 279 and 299 nm also increased gradually [17]. This phenomenon was only found in the case of peptide 2, which indicated a strong and unidirectional orientation between interacting GCP units within a self-assembled aggregate of peptide 2 [22]. In addition, two contiguous negative CD signals at 222 and 206 nm indicated that peptide 2 adopts an α-helix conformation, brought about by an ionic GCP-carboxylate interaction and an additional π-π interaction between the GCP units [23]. Interestingly, peptide 3 shows a different conformation compared to peptide 1 and peptide 2. Due to the low solubility of peptide 3 in water (pH 7.0), we selected a concentration of 0.5 mmol/L as maximum concentration to explore its secondary structure (instead of 1.0 mmol/L, as used for peptides 2 and 3). The CD spectrum of peptide 3 showed characteristic signals for a β-sheet structure (an intense positive band at 198 nm and a negative band at 218 nm) [24]. In summary, these CD results suggest that both the interactions between the GCP units and the length of the peptide backbone play a very important role for the formation of the observed secondary structures.

|
Download:
|
| Fig. 1. CD spectra of (a) peptide 1, (b) peptide 2, (c) peptide 3 at different concentrations at pH 7.0. | |
{kind=link}
Subsequently, transmission electron microscopy (TEM) and atomic force microscopy (AFM) were employed to investigate the further self-assembly process and the morphology of the resulting higher-order nanostructures formed by these peptide amphiphiles in water at pH 7.0. As shown in Figs. 2a and e, the formation of nanoparticles with a diameter of 25 nm was observed for a freshly prepared solution of peptide 1 at a concentration of 0.2 mmol/L by AFM and TEM. Moreover, a small degree of further aggregation could be observed after storing the solution for one week (Figs. 2c and g). However, as the concentration was increased to 1.0 mmol/L, peptide 1 formed fiber-type structures with a length of approximately 1.2 μm (Figs. 2b and f). Furthermore, the fibers possessed an internal structure: As shown in Figs. 2d and h, lefthanded twisted helical fibers with a uniform pitch of 45 nm could be clearly observed. The consistent pitch and helicity indicate that the underlying intermolecular interactions are uniform throughout the whole structure [22]. This phenomenon occurs most likely due to the intrinsic chirality of the constituent L-amino acids, which is in line with the observations in other peptide-based systems [17, 25].

|
Download:
|
| Fig. 2. AFM (a–d) and TEM (e–h) images of peptide 1 at different concentrations at pH 7.0, either freshly prepared or after incubation for one week. | |
{kind=link}
However, peptide 2 exhibited markedly different self-assembly properties (Fig. 3). Though peptide 2 also assembled into small and regular nanoparticles at low concentration (0.2 mmol/L, Figs. 3a and e), increasing the concentration to 1.0 mmol/L led to the formation of bamboo-like nano-assemblies. These are composed of short, rod-like aggregates, which adhere tightly to each other and stretch out to form longer fibers, thus resembling a bamboo-like morphology both in AFM and TEM (Figs. 3b, d, f, h). In this case, we did not find any evidence of morphological changes induced by an aging effect after incubating the sample for one week.

|
Download:
|
| Fig. 3. AFM (a–d) and TEM (e–h) images of peptide 2 at different concentrations at pH 7.0, either freshly prepared or after incubation for one week. | |
{kind=link}
For peptide 3, both TEM and AFM results indicated that solid spherical structures with a diameter around 30 nm were formed at low concentration (0.2 mmol/L, Figs. 4a and e). In contrast, no obvious aging effect was found after an incubation time of one week (Figs. 4c and g). Considering the poor solubility of peptide 3 at 1.0 mmol/L in water (pH 7.0), we selected a concentration of 0.5 mmol/L as high concentration to further explore its self-assembly behavior. Such a freshly prepared sample also exhibited uniform spherical morphology at 0.5 mmol/L. However, a striking aging effect was observed after one week, leading to cross-linking and fusion of the spherical particles into unordered fibrous structures (Fig. 4).

|
Download:
|
| Fig. 4. AFM (a–d) and TEM (e–h) images of peptide 3 at different concentrations at pH 7.0, either freshly prepared or after incubation for one week. | |
{kind=link}
We also studied the self-assembly process of these peptide amphiphiles in solution by means of dynamic light scattering (DLS) (Figs. S13–S15 in Supporting information), which correlated well with the results of AFM and TEM. Additionally, both the conformation of the peptide amphiphiles 1-3 and the subsequent formation of higher-order aggregates were sensitive to pH-changes. Upon adjusting the pH to 3.0, both the GCP and the carboxylate units were protonated, leading to strong intermolecular charge repulsion. Hence, the characteristic CD signal of α-helix or β-sheet structures disappeared and a negative band at 191 nm was detected, which clearly demonstrated that the well-organized α-helix and β-sheet structures transformed into random-coil structures (Fig. S16 in Supporting information). Also, the originally observed self-assembled structures disappeared as shown in TEM images (Fig. 5), probably caused by the missing secondary structure of the amphiphiles in combination with the charge repulsion. Yet, the readjustment of the pH from 3.0 to 7.0 results in the reformation of the original self-assembled structures, demonstrating a full reversibility of the process as expected.

|
Download:
|
| Fig. 5. TEM images of (a) peptide 1 (1.0 mmol/L), (b) peptide 2 (1.0 mmol/L), (c) peptide 3 (0.5 mmol/L) at pH 3.0. | |
{kind=link}
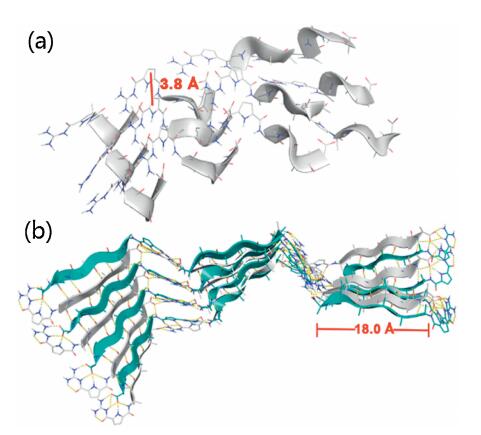
Molecular mechanics calculations (MacroModel, OPLS 2005 force field) were utilized to further explore the underlying intraand intermolecular interactions that lead to the formation of ordered secondary structures from peptide 2 and peptide 3. The energy-minimized α-helix structure formed by peptide 2 showed that the protonated GCP motif not only interacted with the carboxylate group of another peptide, but also with the neighboring GCP-units (Fig. 6a). Thus, a zipper-like interstrand-stacking arrangement of the GCP side chains was realized, which ultimately leads to the formation of a stable α-helix. The average centroid– centroid distance between the pyrrole rings is 3.8 Å (Fig. 6a and Fig. S17 in Supporting information), which is in line with aromatic π-π interactions [26]. Moreover, such close packing and intimate interactions are a possible cause for the bisignate CD signal as described above. For the β-sheet conformation generated for peptide 3, the energy-minimized structure revealed an antiparallel alignment of the strands in β-sheets (Fig. 6b). Such antiparallel orientation allows for the formation of well-aligned hydrogen bonds, so that this superstructure is energetically more favored than the corresponding parallel form [18, 27]. In contrast to the α-helix case, we found two different kinds of GCP-carboxylate interactions: At the termini of the β-sheets, we observed the expected tetradentate GCP-carboxylate interaction involving the guanidinium-group and the pyrrole and amide NH-groups; Within the β-sheet structure, however, only the guanidinium-unit interacts with the neighboring carboxylate in a bidentate fashion. In combination with the extensive hydrogen bonding interactions between the peptide backbones, these interactions are the major driving forces to stabilize the β-sheet structure. The length of the β-sheet backbone formed by peptide 3 is about 18.0 Å (Fig. 6b and Fig. S18 in Supporting information). Notably, the higher hydrophobicity property of peptide 3 may also favor the formation of β-sheets relative to peptide 1 or peptide 2 [18].

|
Download:
|
| Fig. 6. Molecular modeling of (a) α-helix structure formed by peptide 2, (b) β-sheet structure formed by peptide 3 at pH 7.0. | |
{kind=link}
In summary, we developed a series of GCP-containing peptide amphiphiles with different backbone length, which allowed for the controlled formation of different secondary structures, leading to highly diverse morphologies upon further self-assembly. The random coil conformation, α-helix, and β-sheet of peptide 1, peptide 2, and peptide 3, respectively, were clearly proven by CD spectroscopy under neutral conditions. Furthermore, all these peptide amphiphiles aggregated to nanoparticles at low concentrations. At higher concentrations, a left-handed twisted helical fiber with uniform pitch appeared for peptide 1, while peptide 2 gave longer bamboo-like structures, as proven by AFM and TEM. In contrast, incubation of a freshly prepared sample of peptide 3 for one week led to a morphological transition from nanoparticles to disordered fibers. This work provides a simple and useful guideline for the precise control of secondary structures and the resulting higher-order assemblies using peptide amphiphiles. Such a supramolecular strategy is highly beneficial for our in-depth understanding of the self-assembly processes in general.
Declaration of competing interestAll the authors confirm that there are no conflicts to declare.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 21572101), the Natural Science Foundation of Jiangsu Province (No. BK20180055), and Qinghai Provincial Key Laboratory of Tibetan Medicine Research (No. 2017-ZJ-Y11). X.-Y. Hu thanks Prof. Leyong Wang from Nanjing University for the helpful discussion. J. Niemeyer would like to acknowledge funding by the Fonds der Chemischen Industrie (Liebig-Fellowship) and would like to thank Prof. Carsten Schmuck for his support. M. Giese thanks the Professor-Werdelmann Stiftung for generous support.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.036.
| [1] |
A.L. Boyle, D.N. Woolfson, Chem. Soc. Rev. 40 (2011) 4295-4306. DOI:10.1039/c0cs00152j |
| [2] |
H. Acar, S. Srivastava, E.J. Chung, et al., Adv. Drug Deliv. Rev. 110 (2017) 65-79. |
| [3] |
Y. Xu, M. Tian, H. Zhang, et al., Chin. Chem. Lett. 29 (2018) 1093-1097. DOI:10.1016/j.cclet.2018.03.032 |
| [4] |
Y. Sun, A. Zhan, S. Zhou, et al., Chin. Chem. Lett. 30 (2019) 1435-1439. DOI:10.1016/j.cclet.2019.05.001 |
| [5] |
D. Mandal, A.N. Shirazi, K. Parang, Org. Biomol. Chem. 12 (2014) 3544-3561. DOI:10.1039/C4OB00447G |
| [6] |
M.T. Jeena, L. Palanikumar, E. Min Go, et al., Nat. Commun 8 (2017) 26. DOI:10.1038/s41467-017-00047-z |
| [7] |
I.W. Hamley, Chem. Rev. 112 (2012) 5147-5192. DOI:10.1021/cr3000994 |
| [8] |
F. Chiti, C.M. Dobson, Annu. Rev. Biochem. 75 (2006) 333-366. DOI:10.1146/annurev.biochem.75.101304.123901 |
| [9] |
D.J. Selkoe, Nat. Cell Biol. 6 (2004) 1054-1061. DOI:10.1038/ncb1104-1054 |
| [10] |
G. Wei, Z. Su, N.P. Reynolds, et al., Chem. Soc. Rev. 46 (2017) 466-4708. |
| [11] |
Y. Jiang, H. Long, Y. Zhu, Y. Zeng, Chin. Chem. Lett. 29 (2018) 1067-1073. DOI:10.1016/j.cclet.2018.05.028 |
| [12] |
C.J. Bruns, J.F. Stoddart, Nat. Nanotechnol. 8 (2013) 9-10. DOI:10.1038/nnano.2012.239 |
| [13] |
J.C. Stendahl, M.S. Rao, M.O. Guler, S.I. Stupp, Adv. Funct. Mater. 16 (2006) 499-508. DOI:10.1002/adfm.200500161 |
| [14] |
M.P. Hendricks, K. Sato, L.C. Palmer, S.I. Stupp, Acc. Chem. Res. 50 (2017) 2440-2448. DOI:10.1021/acs.accounts.7b00297 |
| [15] |
C. Schmuck, M. Schwegmann, J. Am. Chem. Soc. 127 (2005) 3373-3379. DOI:10.1021/ja0433469 |
| [16] |
J. Hatai, C. Schmuck, Acc. Chem. Res. 52 (2019) 1709-1720. DOI:10.1021/acs.accounts.9b00142 |
| [17] |
X.Y. Hu, M. Ehlers, T. Wang, et al., Chem. -Eur. J. 24 (2018) 9754-9759. DOI:10.1002/chem.201801315 |
| [18] |
M. Rad-Malekshahi, L. Lempsink, M. Amidi, W.E. Hennink, E. Mastrobattista, Bioconjug. Chem. 27 (2016) 3-18. DOI:10.1021/acs.bioconjchem.5b00487 |
| [19] |
C.J.C. Edwards-Gayle, I.W. Hamley, Org. Biomol. Chem. 15 (2017) 5867-5976. DOI:10.1039/C7OB01092C |
| [20] |
C.A. Chantell, M.A. Onaiyekan, M. Menakuru, J. Pept. Sci. 18 (2012) 88-91. DOI:10.1002/psc.1419 |
| [21] |
S.M. Kelly, N.C. Price, Curr. Protein Pept. Sci. 1 (2000) 349-384. DOI:10.2174/1389203003381315 |
| [22] |
P. Jana, M. Ehlers, E. Zellermann, K. Samanta, C. Schmuck, Angew. Chem. Int. Ed. 55 (2016) 15287-15291. DOI:10.1002/anie.201608069 |
| [23] |
E. Yashima, N. Ousaka, D. Taura, et al., Chem. Rev. 116 (2016) 13752-13990. DOI:10.1021/acs.chemrev.6b00354 |
| [24] |
Y. Hu, R. Lin, P. Zhang, et al., ACS Nano 10 (2016) 880-888. DOI:10.1021/acsnano.5b06011 |
| [25] |
H. Cui, T. Muraoka, A.G. Cheetham, S.I. Stupp, Nano Lett. 9 (2009) 945-951. DOI:10.1021/nl802813f |
| [26] |
S. Aravinda, N. Shamala, C. Das, et al., J. Am. Chem. Soc. 125 (2003) 5308-5315. DOI:10.1021/ja0341283 |
| [27] |
A. Perczel, Z. Gáspári, I.G. Csizmadia, J. Comput. Chem. 26 (2005) 1155-1168. DOI:10.1002/jcc.20255 |