 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Pollution Control and Resources Reuse, Shanghai Institute of Pollution Control and Ecological Security, School of Environmental Science and Engineering, Tongji University, Shanghai 200092, China;
c College of Chemical Engineering, Zhejiang University of Technology, Hangzhou 310014, China;
d College of Chemistry and Molecular Engineering, Zhengzhou University, Zhengzhou 450001, China;
e School of Chemistry and Environmental Engineering, Key Laboratory for Green Chemical Process(Ministry of Education), Hubei Key Lab. of Novel Reactor&Green Chemical Technology, Wuhan Institute of Technology, Wuhan 430205, China
Climatic deterioration and energy crisis have aroused global concern in searching for sustainable energy-storage apparatuses [1-3]. Supercapacitors stand out by virtue of the advantageous power density and cycle stability [4-6]. However, boosting the energy storage without sacrificing power/cycling performance is fundamental for their practical applications [7]. Generally, supercapacitors are divided into electric double-layer capacitors based on a physical charge-storage mechanism, and pseudo-capacitors relying on transition metal oxides and conducting polymers with redox activities [8, 9]. Since the former way is purely electrostatic, the capacitance is positively correlated with the quantity of electrolyte ions stored on the electrode surface, and thus large surface area is essential [10, 11]. While the latter delivers pseudocapacitance realized by rapid faradaic reactions, but the long-term stability and power density would be compromised [12-14].
Heteroatom-doped carbon supercapacitors integrate both large adsorbing carbon surfaces and faradaic-active heteroatom species [15, 16]. A high ion-accessible carbon surface is the prerequisite for sufficient electrostatic adsorption sites [17, 18]. For one thing, hierarchical pore distribution is responsible for effective utilization of the surface area to maximize adsorbed ions on electrode materials [11, 19]. This is because micropores contribute to the formation of electric double layers, and larger pores serve as highways to cut down the distance from internal surface to interface [20, 21]. For another, doping heteroatoms (N, P, O, S, etc.) into carbon matrix can break the electroneutrality of neighbored C atoms, thus modifying surface polarization/affinity toward electrolyte for enhanced electrostatic interaction [22-24]. More importantly, faradaic-active species (e.g., pyrrolic N, pyridinic N and quinone O) also yield the pseudocapacitance through redox reactions [25, 26]. Nevertheless, overmuch incorporation of heteroatoms may lead to partial collapses of carbon skeleton and thus decayed cycle stability of the electrode [27]. Hence, an effective approach of synthesizing heteroatom-doped carbons should be conceived to maximize the electrostatic interaction and redox reactions.
In this work, we report a straightforward approach to construct N, O-doped carbons (NOCx, y, where x reflects the pyrolysis temperature and y demonstrates the o-tolidine/ p-benzoquinone molar ratio) composed of the vast adsorbing surface and diverse faradaic-active species. The present synthetic process allows the transfer of parent heteroatom-containing functionalities to the final carbons. The NOCs have a high adsorption surface (1799 m2/g), a multi-level micro–meso–macro porous architecture and abundant N/O functionalities, which lower the contact resistance and additionally impart large pseudocapacitive contribution to the total capacitance (311 F/g). Furthermore, the NOC-assembled two-electrode cell affords high faradaic activity in a redox-active solution and yields a high aqueous energy delivery of 18.0 Wh/kg under the power output of 350 W/kg. By utilizing high-voltage electrolytes, the assembled devices display the high energy density up to 74.9 Wh/kg and long cycling lifetime (87.8% retention after 10, 000 cycles).
The functional groups of o-tolidine/p-benzoquinone polymers (PTBy) are confirmed via the Fourier-transformed infrared (FT-IR) spectra in Fig. S1a (Supporting information). Obvious absorptions at 806, 1281, 1488 and 1562 cm-1 represent N-H, C-N, C=C and C=O groups [16, 28]. X-ray diffraction (XRD) patterns of NOCs (Fig. S1b in Supporting information) show dual broad diffraction peaks at 2θ = 26° and 43°, corresponding to the (002) and (100) diffractions of the amorphous carbonaceous phase [29, 30]. The energy dispersive spectroscopy (EDS) mappings depicted in Fig. S1c (Supporting information) present the homogenous distribution of C, N and O elements within NOCs, demonstrating a successful doping of faradaic-active sites. The thermogravimetric analysis indicates the high carbon yields (48%–64%) of PTBs (Fig. S1d in Supporting information). Owing to the electron-donor effect of the primary diamine (o-tolidine) and the electronwithdrawing capability of the carbonyl groups (p-benzoquinone), o-tolidine and p-benzoquinone polymerize through Michael addition with amino groups connected at the 2, 5-position of p-benzoquinone [31]. During the pyrolysis process, benzenoid rings in the polymer serve as cross-linking moieties to join in the carbon skeleton, while N/O faradaic-active sites supplied by the o-tolidine/p-benzoquinone are gradually anchored into the final matrix.
The wide-scan X-ray photoelectron spectra (XPS) of NOCs (Figs. S2a and b in Supporting information) illustrate the typical N 1s, O 1s and C 1s peaks. As shown in Table S1 (Supporting information), with the rising dosage of N source (o-tolidine), the contents of N atom in NOCs go up from 2.56 at% to 4.35 at% and O atom range from 6.59 at% to 8.54 at%. Meanwhile, higher temperature leads to the decreasing of N/O contents from 4.59/12.20 at% to 1.99/5.76 at%. The N 1s spectra shown in Figs. S3a–c (Supporting information) are fitted into four peaks including N-6 (pyridinic N, 398.2 eV), N-5 (pyrrolic N, 399.5 eV), N–Q (quaternary N, 400.3 eV) and N–X (oxidized N, 401.6 eV), respectively [32, 33]. By deconvoluting the O 1s spectra, three forms of O element corresponding to O-I (quinone O, 530.7 eV), O-Ⅱ (C-OH, 532.4 eV) and O-Ⅲ (O=C-O, 533.6 eV) can be recognized in Figs. S3d–f (Supporting information) [9]. The C 1s spectra (Figs. S3g–i in Supporting information) can be deconvoluted into three types: C——C (284.3 eV), C-N/C-O (285.6 eV) and C=O (287.3 eV), revealing binding situations with N/O species [25]. N/O dopants can break the electroneutrality of neighbored C atoms for a polarized NOC surface, and thus boost the electrode wettability with smaller contact angles (31°–59°) proven in Fig. S4 (Supporting information) [22, 23]. Besides, faradaic-active species (N-6, N-5 and O-I) in both N and O atoms could serve as Lewis base sites in acid electrolyte, and provide pseudocapacitance superiority to the ultimate performance [4, 34]. As shown in Table S2 (Supporting information), regulating molar ratios in the synthesis leads to a flexible content (2.39%–3.88%) of faradaic-active N and O species. Therefore, the o-tolidine/p-benzoquinone molar ratio is significant for adjusting the electrode wettability and faradaic activity for high capacitive performance.
Texture parameters of NOCs are investigated by N2 sorption measurements. The trait of I-type curves (Fig. 1a) in all the NOCs is confirmed by steep uptake of nitrogen (P/P0 < 0.05), manifesting copious micropores [35, 36]. The ascending o-tolidine interspersed in PTBs makes the polymer skeleton fragile and unable to withstand the high-temperature pyrolysis, and the surface area of NOC700, y gradually declines from 1836 m2/g to 1454 m2/g (Table S3 in Supporting information). Besides, a hierarchical architecture in NOC700, 1:1 (Fig. 1b, insert) is advantageous for continuously offering ion-transfer channels, and thus the effect of pyrolysis temperatures is subsequently investigated. As the temperature elevates from 600 ℃ to 700 ℃, the micropores at 0.5 nm partially develop into abundant larger ones (Fig. 1b) with boosted surface area from 1129 m2/g to 1799 m2/g (Table S3). This is because more molecules escape from the carbon matrix during the assisted KOH-activation process. However, excessive pyrolysis temperature (800 ℃) leads to the reduction in micropores centered at 0.5 and 0.8 nm (Fig. 1b) and the decrease of surface area (1128 m2/g, Table S3). The fillings of mesopores in NOCx, 1:1 are confirmed by small hysteresis loops (P/P0 > 0.45) in Fig. 1a [37]. The tendency of mesoporous distribution evolves from the immature texture (NOC600, 1:1) to a developed one (NOC700, 1:1) with more mesopores spreading within 2–10 nm (Fig. 1b, insert) due to the extension/amalgamation of smaller pores with the elevation of heat-treatment temperature [38]. Further increase of temperature results in partial collapses of the carbon framework, and thus leads to the decreased surface area/pore volume for NOC800, 1:1 (Table S3). The macroporosities of NOCs are proven by scanning electron microscope (SEM). The morphologies presented in Figs. S5a–c (Supporting information) indicate the framework of NOCs become more fragile with the rising dosage of N source (o-tolidine). More macropores are observed in NOC700, 1:1 than in NOC600, 1:1 in Figs. S5b and d (Supporting information). Nevertheless, the morphology suffers from structural fracture owing to excessive pyrolysis at 800 ℃ (Fig. S5e in Supporting information). Therefore, optimized molar ratio and pyrolysis temperature would yield adequate surface area and desirable hierarchically coexisting micro–meso–macro structures for double interlayer adsorption and ion diffusion.

|
Download:
|
| Fig. 1. N2 sorption isotherms (a) and pore distributions of NOCs (b). | |
{kind=link}
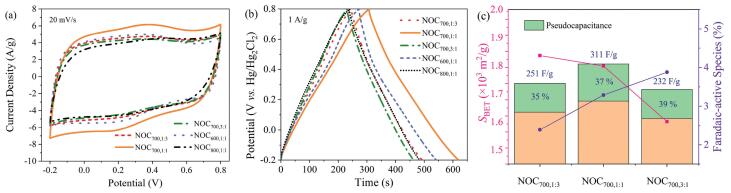
Electrochemical performances of NOCs are first investigated in a three-electrode configuration. Compared with the nearly rectangular cyclic voltammetry (CV) curves and isosceles-triangular shaped galvanostatic charging/discharging (GCD) profiles in the KOH electrolyte (Figs. S6a and b in Supporting information), the extra redox peaks illustrated in Fig. 2a and slight distortions in Fig. 2b verify the occurrence of faradaic reactions by N, O-containing functionalities (N-6, N-5 and O-I) gaining/losing protons in an acidic solution (Fig. S6c in Supporting information) [39-41]. The gravimetric capacitances calculated by the GCD discharging branches are 232/251 (NOC700, 1:3), 290/311 (NOC700, 1:1), 212/232 (NOC700, 3:1), 251/272 (NOC600, 1:1), 209/ 237 F/g (NOC800, 1:1) in KOH/H2SO4 electrolytes, respectively. The optimal NOC700, 1:1 electrode also gives strong rate capability (Figs. S6d and e in Supporting information) with capacitance retention of 66% in KOH and 69% in H2SO4 at 10 A/g. It can be found that as the pyrolysis temperature rises from 600 ℃ to 700 ℃, the capacitances exhibit an obvious improvement (Fig. 2b). This is mainly because sufficient pyrolysis at 700 ℃ guarantees a developed porous architecture equipped with better conductivity and lower contact resistance, which can be validated by electrochemical impedance spectrogram (EIS) plots (Fig. S6f in Supporting information). The ohmic resistance (Rs) deduced by the first Z' intersection is only 0.25 Ω for NOC700, 1:1, suggesting that the electrode possesses a low internal/contact resistance [42]. Moreover, the small charge-transfer resistance (Rct) value (0.84 Ω) and the Warburg impedance (W, 0.45 Ω) can guarantee the superb propagation capability for ions within NOC700, 1:1 [43]. Further increase of pyrolysis temperature to 800 ℃ brings about decayed utilization of adsorbing surface, which explains the unsatisfactory capacitance of NOC800, 1:1 in Fig. 2b. Meanwhile, faradaic-active species regulated by o-tolidine/p-benzoquinone molar ratios endow the electron-donor features and supply abundant electrochemically active sites. To better confirm the pseudocapacitive contributions, the relationship between specific capacitance and square root of charge/discharge time is plotted in Fig. S6g (Supporting information). The Y-intercepts account for the rate-independent parts, while the remainders of total capacitances are pseudocapacitances [44]. The correlation between capacitances, surface areas and faradaic-active heteroatom species is given in Fig. 2c. With the increasing faradaic-active heteroatom species, the pseudocapacitive contribution is evidenced by a scrambling tendency from 35% to 39%. However, excessive introduction of N-containing dopants leads to a decayed surface area of the flimsy carbon matrix for physical adsorption, and renders NOC700, 3:1 with inferior capacitive performance. Therefore, NOC700, 1:1 takes full use of the integrated superiorities from adsorbing surface and faradaic-active sites, obtaining a maximized capacitance.

|
Download:
|
| Fig. 2. CV/GCD curves (a, b) of NOCs in 1 mol/L H2SO4. The relationships between capacitances and surface area/faradaic-active species (c). | |
{kind=link}
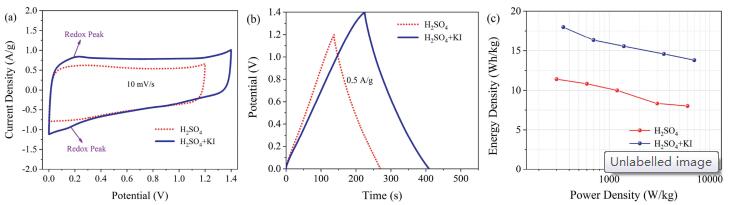
Regarding practical applications, NOC700, 1:1 electrodes are assembled into a symmetric cell with the redox-active hybrid electrolyte H2SO4+KI. The larger CV integral area in H2SO4+KI (Fig. 3a) originates from the four additional redox couples (3I-/I3-, 2I-/I2, 2I3-/3I2 and I2/2IO3-). The reversible reactions between them contribute to the extra paired redox peaks providing additional pseudocapacitance [45-47]. Additionally, the similar standard potential of redox couples causes O2 evolution potential shift, hindering H2O electrolysis and enlarging the device potential from 1.2 V to 1.4 V [45, 48]. The CV curves display highly symmetric rectangular profiles and maintain nearly rectangle even at 100 mV/s (Fig. S7a in Supporting information) due to the outstanding ion sorption reversibility [49]. As exhibited in GCD profiles (Fig. 3b), the capacitance performance of NOC700, 1:1 reaches a higher level of 264 F/g @ 0.5 A/g in H2SO4 + KI which is 1.18-fold reinforcement than in H2SO4 (228 F/g). The small Rct and Rs values (1.91 and 0.47 Ω for H2SO4+KI cell, and 1.46 and 0.19 Ω for H2SO4 one) indicate low internal resistances (Fig. S7b in Supporting information). Highenergy storage is achieved benefited from the double-win of enlarged voltage window (1.2 V–1.4 V) and enhanced capacitance generated by redox-active electrolyte. The faradaic-active cell exhibits energy exports of 18.0 Wh/kg at the power source of 350 W/kg and maintains 13.8 Wh/kg @ 7000 W/kg in H2SO4 + KI, enhancing greatly compared with the energy/power properties in H2SO4 (11.4 Wh/kg @ 300 W/kg and 8.0 Wh/kg @ 6000 W/kg, Fig. 3c). Besides, the device shows an outstanding electrochemical life over a consecutive charging-discharging process. Notably, considerable capacitance enhancement by the enlarged faradic effect renders the performancehigher inH2SO4+KI (199 F/g) than in H2SO4 (188 F/g) even after 10, 000 cycles (Fig. S7c in Supporting information). Therefore, N, O-containing species furnish faradaicactive sites to react with redox pairs in H2SO4 + KI, thus feasible for energy storage in terms of both the device potential and pseudocapacitive activity.

|
Download:
|
| Fig. 3. Comparison of CV/GCD curves (NOC700, 1:1) in a two-electrode H2SO4 + KI device with H2SO4 electrolyte (a, b) and Ragone plots (c). | |
{kind=link}
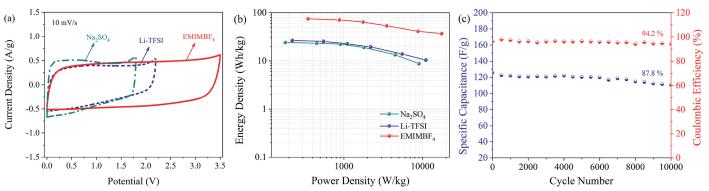
Due to the enhanced electrochemical behavior caused by a superior ion-accessible platform and an ideal nanoarchitecture, the NOC700, 1:1 electrodes are subsequently fabricated into symmetric cells using 1 mol/L Na2SO4, 7 mol/kg LI-TFSI and EMIMBF4. The quasi-rectangular/triangular shaped CV/GCD profiles (Fig. 4a and Figs. S8a–c in Supporting information) illustrate high efficiency in the reversible charge-storage process [50]. Compared with Na2SO4-loaded symmetric device, the LI-TFSI and EMIMBF4 assembled capacitors show higher Rct values disclosed in Fig. S8d (Supporting information) owing to larger ionic sizes (TFSI-: 0.79 nm, EMIM+: 0.76 nm, BF4-: 0.48 nm) [51]. The assembled Na2SO4/LI-TFSI/EMIMBF4 cells with significantly broadened potentials offer boosted energy outputs of 24.1/26.5/74.9 Wh/kg (Fig. 4b). Besides, over 10, 000 cycles at 2 A/g, the capacitance in EMIMBF4 only decays by 12.2% with 94.2% Coulombic efficiency retaining (Fig. 4c). The NOC-based devices achieve distinguished energy deliveries (Table S4 in Supporting information), advantageous power outputs and prominent cycle stabilities for advanced supercapacitors. Firstly, a high adsorbing surface (1799 m2/g) promotes physical charge accumulation [17, 18]. Secondly, the hierarchical porous texture leads to effective utilization of the carbon surface, in favor of ion-adsorption on the electrode material [11, 52]. Thirdly, by doping N/O into carbon matrix, the surface polarization/affinity toward electrolyte can be modified for enhanced electrostatic interaction and fast ion transfer at high rates [22, 53]. Besides, the faradaic-active functionalities stem from heteroatoms also yield pseudocapacitive contributions [25].

|
Download:
|
| Fig. 4. Two-electrode electrochemical tests of NOC700, 1:1 using 1 mol/L Na2SO4, 7 mol/kg Li-TFSI and EMIMBF4: Comparison of CV curves (a), Ragone plots (b), pattern of Coulombic efficiency and cycling test at 2 A/g in EMIMBF4 (c). | |
{kind=link}
To conclude, the in-situ polymerization/pyrolysis approach is employed to efficiently construct highly active N, O-doped carbons (NOCs), which offers diversified electrochemically active sites, a vast surface area and hierarchical porous texture. A variety of faradaic-active species (N-6, N-5 and O-I) are homogeneously distributed on NOC architecture. Besides, the high adsorption platform (1799 m2/g) and multi-level channels are optimized for electrostatic accumulation and continuous ion-transmission. Consequently, a 37% pseudocapacitive contribution and a corresponding maximized capacitance (311 F/g) are obtained by the asprepared NOC electrode. A reinforced energy delivery (18.0 Wh/kg) at a higher power output (350 W/kg) is achieved via the electron gaining/losing process of the redox-pairs in the aqueous faradaicactive cell. The NOC700, 1:1-assembled capacitor also possesses a competitive energy density (74.9 Wh/kg) and high-potential stability (87.8% retention after 10, 000 cycles) in EMIMBF4. We believe this in-situ polymerization/pyrolysis approach will open up an avenue toward ameliorating both faradaic activity and nanoarchitecture for promising energy-storage apparatuses.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was financially supported by the National Natural Science Foundation of China (Nos. 21905207, 21875165, 51772216, and 21703161), the Science and Technology Commission of Shanghai Municipality, China (No. 14DZ2261100), and the Natural Foundation of Hubei Province of China (No. 2014CFB782) and the Fundamental Research Funds for the Central Universities.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2020.02.026.
| [1] |
S. Choudhury, S. Stalin, D. Vu, et al., Nat. Commun. 10 (2019) 4398. DOI:10.1038/s41467-019-12423-y |
| [2] |
H.G. Jung, J. Hassoun, J.B. Park, et al., Nat. Chem. 4 (2012) 579-585. DOI:10.1038/nchem.1376 |
| [3] |
F. Wang, X. Wu, X. Yuan, et al., Chem. Soc. Rev. 46 (2017) 6816-6854. DOI:10.1039/C7CS00205J |
| [4] |
W. Zhang, C. Xu, C. Ma, et al., Adv. Mater. 29 (2017) 1701677. DOI:10.1002/adma.201701677 |
| [5] |
C. Peng, J. Yu, S. Chen, L. Wang, Chin. Chem. Lett. 30 (2019) 1137-1140. DOI:10.1016/j.cclet.2019.02.007 |
| [6] |
S. Witomska, Z. Liu, W. Czepa, et al., J. Am. Chem. Soc. 141 (2019) 482-487. DOI:10.1021/jacs.8b11181 |
| [7] |
F. Wei, X. He, H. Zhang, et al., J. Power Sources 428 (2019) 8-12. DOI:10.1016/j.jpowsour.2019.04.096 |
| [8] |
A. González, E. Goikolea, J.A. Barrena, R. Mysyk, Renew. Sustain. Energy Rev. 58 (2016) 1189-1206. DOI:10.1016/j.rser.2015.12.249 |
| [9] |
Y. Liao, H. Wang, M. Zhu, A. Thomas, Adv. Mater. 30 (2018) 1705710. DOI:10.1002/adma.201705710 |
| [10] |
C. Wang, D. Wu, H. Wang, et al., J. Mater. Chem. A 6 (2018) 1244-1254. DOI:10.1039/C7TA07579K |
| [11] |
H. Zhong, F. Xu, Z. Li, et al., Nanoscale 5 (2013) 4678-4682. DOI:10.1039/c3nr00738c |
| [12] |
S.K. Das, K. Bhunia, A. Mallick, et al., Microporous Mesoporous Mater. 266 (2018) 109-116. DOI:10.1016/j.micromeso.2018.02.026 |
| [13] |
B. You, L. Wang, L. Yao, J. Yang, Chem. Commun. 49 (2013) 5016-5018. DOI:10.1039/c3cc41949e |
| [14] |
J. Zhu, Y. Dong, S. Zhang, Z. Fan, Acta Phys. -Chim. Sin. 36 (2020) 1903052. DOI:10.3866/PKU.WHXB201903052 |
| [15] |
S.S. Karade, B.R. Sankapal, ACS Sustain. Chem. Eng. 6 (2018) 15072-15082. DOI:10.1021/acssuschemeng.8b03366 |
| [16] |
M.P. Chavhan, S. Ganguly, Carbon 154 (2019) 33-41. DOI:10.1016/j.carbon.2019.07.091 |
| [17] |
J. Zhao, H. Lai, Z. Lyu, et al., Adv. Mater. 27 (2015) 3541-3545. DOI:10.1002/adma.201500945 |
| [18] |
D. Xue, D. Zhu, H. Duan, et al., Chem. Commun. 55 (2019) 11219-11222. DOI:10.1039/C9CC06008A |
| [19] |
N. Fu, J. Yu, J. Zhao, et al., Carbon 149 (2019) 538-545. DOI:10.1016/j.carbon.2019.04.076 |
| [20] |
W. Lei, J. Guo, Z. Wu, et al., Sci. Bull. 62 (2017) 1011-1017. DOI:10.1016/j.scib.2017.06.001 |
| [21] |
Y. Lu, J. Liang, S. Deng, et al., Nano Energy 59 (2019) 103993. |
| [22] |
P. Sazama, J. Pastvova, C. Rizescu, et al., ACS Catal. 8 (2018) 1779-1789. DOI:10.1021/acscatal.7b04086 |
| [23] |
H. Zhou, Y. Zhou, L. Li, et al., ACS Sustain. Chem. Eng. 7 (2019) 9281-9290. DOI:10.1021/acssuschemeng.9b00279 |
| [24] |
L. Sun, H. Zhou, L. Li, et al., ACS Appl. Mater. Interfaces 9 (2017) 26088-26095. DOI:10.1021/acsami.7b07877 |
| [25] |
L. Miao, H. Duan, Z. Wang, et al., Chem. Eng. J. 382 (2020) 122945. DOI:10.1016/j.cej.2019.122945 |
| [26] |
M. Zhang, C. Yu, Z. Ling, et al., Green Chem. 21 (2019) 2095-2103. DOI:10.1039/C9GC00655A |
| [27] |
W.J. Lee, J. Lim, S.O. Kim, Small Methods 1 (2017) 1600014. DOI:10.1002/smtd.201600014 |
| [28] |
R. Rajak, M. Saraf, S.M. Mobin, J. Mater, Chem. A 7 (2019) 1725-1736. |
| [29] |
L. Miao, X. Qian, D. Zhu, et al., Chin. Chem. Lett. 30 (2019) 1445-1449. DOI:10.1016/j.cclet.2019.03.010 |
| [30] |
W. Li, B. Li, M. Shen, et al., Chem. Eng. J. 384 (2020) 123309. DOI:10.1016/j.cej.2019.123309 |
| [31] |
B.D. Mather, K. Viswanathan, K.M. Miller, T.E. Long, Prog. Polym. Sci. 31 (2006) 487-531. DOI:10.1016/j.progpolymsci.2006.03.001 |
| [32] |
Z. Song, H. Duan, D. Zhu, et al., J. Mater. Chem. A 7 (2019) 15801-15811. DOI:10.1039/C9TA02690H |
| [33] |
L. Li, Y. Zhou, H. Zhou, et al., ACS Sustain. Chem. Eng. 7 (2018) 1337-1346. |
| [34] |
Y. Dong, W. Wang, H. Quan, et al., ChemElectroChem 3 (2016) 814-821. DOI:10.1002/celc.201500519 |
| [35] |
J. Yan, D. Zhu, Y. Lv, et al., Chin. Chem. Lett. 31 (2020) 579-582. DOI:10.1016/j.cclet.2019.05.035 |
| [36] |
F. Wei, H. Bi, S. Jiao, X. He, Acta Phys. -Chim. Sin. 36 (2020) 1903043. DOI:10.3866/PKU.WHXB201903043 |
| [37] |
Y. Li, G. Wang, T. Wei, et al., Nano Energy 19 (2016) 165-175. DOI:10.1016/j.nanoen.2015.10.038 |
| [38] |
Y. Liu, J. Cao, X. Jiang, et al., J. Mater. Chem. A 6 (2018) 6891-6903. DOI:10.1039/C8TA01155A |
| [39] |
X. Yu, C. Pei, L. Feng, Chin. Chem. Lett. 30 (2019) 1121-1125. DOI:10.1016/j.cclet.2019.01.009 |
| [40] |
H. Li, J. Li, A. Thomas, Y. Liao, Adv. Funct. Mater. 29 (2019) 1904785. DOI:10.1002/adfm.201904785 |
| [41] |
H.A. Andreas, B.E. Conway, Electrochim. Acta 51 (2006) 6510-6520. DOI:10.1016/j.electacta.2006.04.045 |
| [42] |
F. Sun, Z. Qu, J. Gao, et al., Adv. Funct. Mater 28 (2018) 1804190. DOI:10.1002/adfm.201804190 |
| [43] |
J. Yu, C. Yu, W. Guo, et al., Nano Energy 64 (2019) 103921. DOI:10.1016/j.nanoen.2019.103921 |
| [44] |
J.S.M. Lee, M.E. Briggs, C.C. Hu, A.I. Cooper, Nano Energy 46 (2018) 277-289. DOI:10.1016/j.nanoen.2018.01.042 |
| [45] |
B. Nagar, D.P. Dubal, L. Pires, et al., ChemSusChem 11 (2018) 1849-1856. DOI:10.1002/cssc.201800426 |
| [46] |
C. Zhong, Y. Deng, W. Hu, et al., Chem. Soc. Rev. 44 (2015) 7484-7539. DOI:10.1039/C5CS00303B |
| [47] |
K. Jayaramulu, D.P. Dubal, B. Nagar, et al., Adv. Mater. 30 (2018) 1705789. DOI:10.1002/adma.201705789 |
| [48] |
S.J. Yoo, B. Evanko, X. Wang, et al., J. Am. Chem. Soc. 139 (2017) 9985-9993. DOI:10.1021/jacs.7b04603 |
| [49] |
C. Chen, G. Xu, X. Wei, L. Yang, J. Mater. Chem. A 4 (2016) 9900-9909. DOI:10.1039/C6TA04062D |
| [50] |
L. Sun, Y. Zhou, L. Li, et al., Appl. Surf. Sci. 467 (2019) 382-390. |
| [51] |
P. Simon, Y. Gogotsi, Nat. Mater. 7 (2008) 845-854. DOI:10.1038/nmat2297 |
| [52] |
M. Fu, W. Chen, X. Zhu, et al., Carbon 141 (2019) 748-757. DOI:10.1016/j.carbon.2018.10.034 |
| [53] |
X. Qian, L. Miao, J. Jiang, et al., Chem. Eng. J. 388 (2020) 124208. DOI:10.1016/j.cej.2020.124208 |