 2020, Vol. 31
2020, Vol. 31 


b University of Chinese Academy of Sciences, Beijing 100049, China
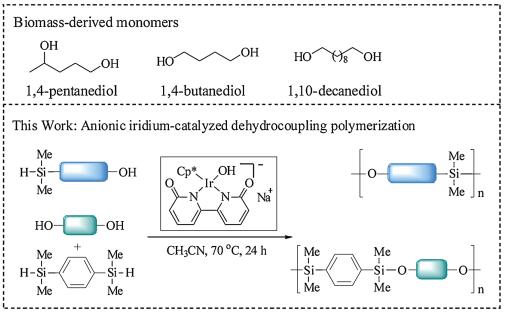
Due to the programmed depletion of fossil resources, nonre-newable and nondegradable synthetic polymers produced from fossil resources pose a serious threat to the ecosystem [1, 2]. It is beneficial to develop polymeric materials sourced from renewable feedstocks [3-8]. Although a number of groups have studied the use of renewable resources-based monomers for the synthesis of polymeric materials [9, 10]. The total volume of polymeric materials synthesized from nonrenewable fossil fuel resources far outweighs that from renewable biomass [11, 12]. The main reason is largely due to the renewable polymeric materials possess high cost and inferior performance compared with polymers produced from petroleum chemicals [13, 14].
Great efforts have been spent on the developing novel and promising renewable feedstocks, and applying these compounds to the synthesis of biopolymers, bioresins, and various value-added chemicals [15-18]. Some of the widely studied renewable feed-stocks are: lactic acid, triglycerides fatty acids, 5-hydroxymethyl-furfural [19] or its derivatives [20, 21], and vanillin. So far, several polymers based on these feedstocks have been developed, including the poly(lactic acid) [22], polyvanillin [23, 24], poly-(hydroxylalkanoate)s, etc. In 2004, the concept of "Top Value Added Chemicals from Biomass" was proposed by the US Department of Energy (DOE), which pointed to clear direction for development of renewable polymeric materials [25, 26].
Among these renewable polymers, PSEs are an important class of polymers with unique and significant properties, for instance, low Tg, good thermal stability, biocompatibility and high gas permeability [27, 28]. These polymers have been applied in many fields, such as high-temperature elastomers [29-31], conductive polymeric mate-rials [32] and chiral column packing materials [33]. As a promising degradable material, PSEs has been studied for decades. Dehy-drocoupling polymerization is one of the most important methods to access PSEs [34]. Different types of monomers have been applied to the synthesis of PSEs [27, 35]. The most commonly used monomers are AA type monomers (dihydroxyl compound) and BB type monomers (silanes). Various dihydroxyl compounds are suitable substrates for the synthesis of PSEs, including diols [36], diphenols [37], water [38] and disilanols [39]. However, most of PSEs are derived from nonrenewable feedstocks, which is the key obstacle to its sustainable development. After analyzing functional group structure of the top 10 chemicals derived from biomass, we found that more than half of these compounds can be transformed to diols via single reduction step with high yields, including succinic acid, 2-hydroxymethyl-5-furfural (HMF), levulinic acid, etc. [40]. These chemicals can be transformed to 1, 4-butanediol, 2, 5-furandimethanol, 1, 4-pentanediol, etc., respectively. Driven by these considerations, we envisaged that applying these diols derived from biomass to the synthesis of PSEs.
To our knowledge, HMF based PSEs have been reported [41]. However, monomers derived from levulinic acid have not been applied to the synthesis of PSEs via dehydrocoupling. As an effective and highly atom-economic method to construct Si-O bond, dehydrocoupling reaction have been catalyzed by transition metal complexes such as palladium [36-38, 42], platinum [38], rhodium [37, 43-45] and manganese [41, 46, 47]. Recently, we reported the synthesis of degradable PSEs from AB-type silyl alcohol monomers catalyzed by homogeneous iridium(I) com-plexes bearing a bisphosphine ligand [48]. Combining our interests in exploring the application of catalytic system based on iridium in dehydrogenative coupling reaction, herein, we reported an anionic iridium complex catalyzed different types (AB type or AA and BB type) of monomers to give high-molecular-weight PSEs (Scheme 1).

|
Download:
|
| Scheme 1. Catalytic dehydrocoupling polymerization for the synthesis of partially biobased PSEs. | |
1, 4-Bis(dimethylsilyl)benzene was obtained from Energy Chemical (Shanghai, China) and distilled from calcium hydride. 1, 4-Butanediol was purchased from Aladdin Reagent Co., Ltd. (Shanghai, China) and dried by azeotropic distillation of toluene and ethanol. 1, 10-Decanediol was purchased from J & K Scientific Ltd. (Beijing, China) and recrystalized from ethyl acetate. Other commercially available reagents were used without further purification. Solvents were treated prior to use according to the standard methods.
To an oven-dried 25 mL resealable Schlenk flask equipped with a magnetic stir bar was charged Ir Cat. (2.8 mg, 0.005 mmol) and acetonitrile (1.5 mL) under nitrogen. The solution was stirred at room temperature for 2 min. For AB type monomers, monomer 1a – 1e (0.5 mmol) was added; for AA and BB type monomers, 1, 4-bis-(dimethylsilyl)benzene (0.5 mmol) and diols 1f–1h (0.5 mmol) were added. The flask was heated at 70 ℃ for 24 h under nitrogen (connected to a nitrogen Schlenk line). During the last 6 h of the reaction time, H2 produced during the reaction was replaced with nitrogen every 2 h. After the polymerization, the reaction mixture was cooled to room temperature, and the content was purified by the precipitation method.
All of the polymers are soluble in dichloromethane (DCM) and insoluble in methanol (MeOH). So, these two solvents were used in the precipitation process. The reaction mixture was first homoge-nized by the addition of as low as possible amount of DCM (1– 2 mL), then cold MeOH was added portionwise (15–20 mL) until it turned to a biphasic mixture. The top layer was taken out, and the bottom viscous/solid layer was washed with MeOH two times until it gave a white/light yellow color viscous/solid polymer. The resulting polymer was dried to a constant weight and character-ized by 1H NMR, 13C NMR, GPC, TG and DSC.
(4-(3-(Dimethylsilyl)propoxy)-phenyl)methanol (1a) was cho-sen as the model monomer to conduct the condition optimization.The results were depicted in Table 1. According to the conditions in our previous work [23], the reaction was performed under neat condition at 80 C. However, only middle-Mn polymer was obtained (entry 1). In order to improve the Mn and Ð of the product, the effects of solvent were conducted. Use tetrahydrofu-ran (THF) or toluene as solvent led to low-Mn polymers (entries 2 and 3). Reactions carried out in 1, 4-dioxane resulted in no product, suggesting that the coordination ability of cyclic ethers might be the main reason for the low-Mn observed with THF (entries 3 and 4). Then, acetonitrile (CH3CN) was used as solvent; a high-molecular-weight polymer with Mn of 14600 was obtained (entry 5). Afterwards, the effects of reaction temperature were performed carefully. Rising temperature led to polymers with lower Mn and broader Ð (entries 6–8). However, lowering the temperature to 60 ℃ resulted in apparent decrease of yield with slightly rising of Mn (entry 10). When reaction temperature was lowered to 70 ℃, a high molecular weight polymer with narrow Ð was obtained albeit with slightly lower yield (entry 9).
|
|
Table 1 Optimization of the reaction conditions.a |

Next, the effects of catalyst loading, CH3CN loading and reaction time were conducted. The results were depicted in Table 1. Increasing or reducing catalyst loading resulted in apparent decrease of PSEs yields (entries 11 and 12). When solvent loading was added to 3.00 mL, polymer with broader Ð was obtained (entry 13). In order to achieving high Mn and narrow Ð, the solvent was reduced to 0.75 mL (entry 14). When reaction time was prolonged to 48 h and 72 h, polymers with high Mn were given, nevertheless, the yields of PSEs droped to 50% and 41%, respectively (entries 15 and 16). Therefore, the optimal reaction conditions were finally established as: cat. (1 mol%)/CH3CN (1.5 mL)/70 ℃/24 h.
With the optimal reaction conditions in hand, the scope and generality of the dehydrocoupling polymerization were next evaluated (Table 2). In the first instance, four AB-type silyl alcohol monomers were examined. Gratifyingly, all the substrates afforded high molecular weight and moderate to high yields (entries 1–5). Furthermore, these polymers were obtained with similar Ð (around 1.80 Ð values).
|
|
Table 2 Substrate scope: AB-type monomers.a |

For the AA and BB type monomers, dehydrocoupling polymeri-zation can also proceeded smoothly (Table 3). We used a commercially available hydrosilane (1, 4-bis(dimethyl-silyl)ben-zene) as AA type monomer. Three diols derived from biomass were chosen as BB type monomers, including 1, 4-pentanediol (1f), 1, 4-butanediol (1g) and 1, 10-decanediol (1h). Among these diols, monomer 1f and 1g were derived from levulinic acid (LA) and succinic acid (SA) independently [25, 26]. When these two diols were applied to the dehydrocoupling polymerization, PSEs 2f and 2g were obtained with high Mn and moderate yields (entries 1 and 2). Monomer 1h was derived from sebacic acid, a principal component of castor oil [49]. When 1h was used, a high-molecular-weight polymer with Mn of 43800 was obtained (entry 3).
|
|
Table 3 Substrate scope: AA and BB type monomers.a |

{kind=link}
To further demonstrate the utility of our methodology, a gram scale reaction was conducted, as shown in Scheme S4 (Supporting information). When monomer 3 (3 mmol) and 1h (3 mmol) were used, a polymer with Mn of 32400 was provided in 87% yield. The Mn was slightly lower than that of the polymerization on small scale. Moreover, pre-polymerization step was also tried, as shown in Scheme S5 (Supporting information). When monomer 3 and 1h were used in the ratio of 1:1.2, both ends of the product 2j (Mn = 11400, Ð = 1.45) should be terminated by OH groups. When the ratio was 1.2:1, both ends of the product 2k (Mn = 6900, Ð = 1.60) should be terminated by SiH groups. The further polymerization employed prepolymers 2j and 2k, providing polymer 2l with higher Mn (yield = 51%, Mn = 17800).
After purification by precipitation, the PSEs produced by the Ir-catalyzed dehydrocoupling polymerization were, in general, color-less/faint yellow, viscous oils/soft solids, depending on the molar mass and backbones of polymers. Thermal properties of polymers 2f and 2h were investigated under nitrogen atmosphere using TGA and DSC, as shown in Figs. S23-S26 (Supporting information). For polymers 2f and 2h, T50 values were kept at around 490 ℃, which indicated these polymers exhibit good thermal stability. However, the T5 value of 2h was higher than 2f. For polymer 2f, low glass transition temperatures (-49.6 ℃) could be detected. Polymer 2h exhibited indiscernible glass transition via DSC.
To investigate the mechanism, experiments about the generation of Ir-H species and its application in dehydrocoupling polymeriza-tion were conducted, as shown in Schemes S6 and S7 (Supporting information). Firstly, the reaction of catalyst and 1, 1, 3, 3-tetra-methyldisiloxane was conducted. The Ir-H species was detected with almost 50% NMR yield [50]. Then, we applied this mixture to the dehydrocoupling of monomer 1a. A high-Mn polymer was obtained (Mn = 16700, yield = 68%). The Mn and yield of PSEs were similar to the aforementioned result under optimal reaction conditions (Mn = 14900, yield = 67%). This result indicated the generation of Ir-H species may occur during the polymerization. Then, a polymerization process under room temperature was conducted. After 6 h, an orange-red flocculent precipitate was observed. NMR spectrum of this orange-red precipitate indicated that part of the catalyst was transformed to Ir-H species. These results strongly suggested the involvement of Ir-H species in the catalytic cycle.
Based on these results and early studies [50], a plausible mechanism was proposed for the iridium-catalyzed dehydrocou-pling polymerization, as shown in Scheme S8 (Supporting information). Firstly, Ir-H species B is formed via hydrolysis reaction of hydrosilane with iridium catalyst A. Then, Ir-H species B undergoes s bond metathesis with diols to give intermediate C accompanied by evolution of hydrogen. Finally, intermediate C reacts with silane to furnish the dehydrocoupling product and regenerates Ir-H species B to complete the catalytic cycle.
As shown in Figs. S3-S6 (Supporting information), the kinetics of polymerization under 10 ℃ and 0 ℃ was performed. At 10 ℃, the reaction conversion (x) increased quickly. Even under 0 ℃, reaction conversion reached 27% within 3 h 10 min. According to these time courses at different temperatures, the relation between ln[1/(1-x)] and reaction time is found to be in linear. And the calculated reaction rate constants are 0.1051 h -1 and 0.1915 h -1 at 0 ℃ and 10 ℃, respectively. According to these data, the calculated apparent activation energy of polymerization in the temperature range of 0– 10 ℃ is about 38.6 kJ/mol.
In conclusion, we have demonstrated that an anionic iridium complex can effectively catalyze dehydrocoupling polymerization. Various PSEs were synthesized from different types (AB type or AA and BB type) of monomers. Importantly, monomers 1e–1h were derived from renewable feedstocks, including rincinoleic acid, levulinic acid (LA), succinic acid (SA) and sebacic acid. A plausible mechanism via an Ir-H species for the polymerization is presented. The results of kinetic experiments indicated that the apparent activation energy in the temperature range of 0–10 ℃ is about 38.6 kJ/mol. Further work will focus on the synthesis of optically active PSEs.
AcknowledgmentsFinancial support from the National Natural Science Foundation of China (No. 21690074), Dalian Bureau of Science and Technology (No. 2016RD07) and Chinese Academy of Sciences (No. XDB17020300) is acknowledged.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.017.
| [1] |
F. van der Ploeg, J Econ, Lit. 49 (2011) 366-420. DOI:10.1257/jel.49.2.366 |
| [2] |
J.R. Jambeck, R. Geyer, C. Wilcox, et al., Science 347 (2015) 768-771. DOI:10.1126/science.1260352 |
| [3] |
K. Yao, C. Tang, Macromolecules 46 (2013) 1689-1712. DOI:10.1021/ma3019574 |
| [4] |
A. Gandini, T.M. Lacerda, A.J.F. Carvalho, E. Trovatti, Chem. Rev. 116 (2016) 1637-1669. DOI:10.1021/acs.chemrev.5b00264 |
| [5] |
I. Delidovich, P.J.C. Hausoul, L. Deng, et al., Chem. Rev. 116 (2016) 1540-1599. DOI:10.1021/acs.chemrev.5b00354 |
| [6] |
Y. Zhu, C. Romain, C.K. Williams, Nature 540 (2016) 354-362. DOI:10.1038/nature21001 |
| [7] |
A. Llevot, P.K. Dannecker, M. von Czapiewski, et al., Chem.-Eur. J. 22 (2016) 11510-11521. DOI:10.1002/chem.201602068 |
| [8] |
D.K. Schneiderman, M.A. Hillmyer, Macromolecules 50 (2017) 3733-3749. DOI:10.1021/acs.macromol.7b00293 |
| [9] |
A. Gandini, T.M. Lacerda, Prog. Polym. Sci. 48 (2015) 1-39. DOI:10.1016/j.progpolymsci.2014.11.002 |
| [10] |
J.D. Badia, O. Gil-Castell, A. Ribes-Greus, Polym. Degrad. Stabil. 137 (2017) 35-57. DOI:10.1016/j.polymdegradstab.2017.01.002 |
| [11] |
C.K. Williams, M.A. Hillmyer, Polym. Rev. 48 (2008) 1-10. DOI:10.1080/15583720701834133 |
| [12] |
G.W. Huber, S. Iborra, A. Corma, Chem. Rev. 106 (2006) 4044-4098. DOI:10.1021/cr068360d |
| [13] |
E.S. Beach, Z. Cui, P.T. Anastas, Energy Environ. Sci. 2 (2009) 1038-1049. DOI:10.1039/b904997p |
| [14] |
P. Anastas, N. Eghbali, Chem. Soc. Rev. 39 (2010) 301-312. DOI:10.1039/B918763B |
| [15] |
P.F. Koha, T.P. Loh, Green Chem. 17 (2015) 3746-3750. DOI:10.1039/C5GC00900F |
| [16] |
F. Hu, J.J. La Scala, J.M. Sadler, G.R. Palmese, Macromolecules 47 (2014) 3332-3342. DOI:10.1021/ma500687t |
| [17] |
A. Bohre, S. Dutta, B. Saha, M.M. Abu-Omar, ACS Sustain. Chem. Eng. 3 (2015) 1263-1277. DOI:10.1021/acssuschemeng.5b00271 |
| [18] |
H. Xia, S. Xu, X. Yan, X. Zuo, Fuel Process. Technol. 152 (2016) 140-146. DOI:10.1016/j.fuproc.2016.06.030 |
| [19] |
A. Gandini, Polym. Chem. 1 (2010) 245-251. DOI:10.1039/B9PY00233B |
| [20] |
A. Gandini, A.J.D. Silvestre, C.P. Neto, A.F. Sousa, M. Gomes, J. Polym. Sci. Part A:Polym. Chem. 47 (2009) 295-298. DOI:10.1002/pola.23130 |
| [21] |
A. Gandini, Macromolecules 41 (2008) 9491-9504. DOI:10.1021/ma801735u |
| [22] |
T. Maharana, S. Pattanaik, A. Routaray, N. Nath, A.K. Sutar, React. Function. Polym. 93 (2015) 47-67. DOI:10.1016/j.reactfunctpolym.2015.05.006 |
| [23] |
M. Firdaus, M.A.R. Meier, Europ. Polym. J. 49 (2013) 156-166. DOI:10.1016/j.eurpolymj.2012.10.017 |
| [24] |
A.S. Amarasekara, B. Wiredu, A. Razzaq, Green Chem. 14 (2012) 2395-2397. DOI:10.1039/c2gc35645g |
| [25] |
T. Werpy, G. Petersen, A. Aden, et al., Top Value AddedChemicals fromBiomass. Volume 1-Results of Screening for Potential Candidates from Sugars and Synthesis Gas, DTIC Document (2004). |
| [26] |
A. Mukherjee, M.J. Dumont, V. Raghavan, Biomass Bioenerg. 72 (2015) 143-183. DOI:10.1016/j.biombioe.2014.11.007 |
| [27] |
Y. Li, Y. Kawakami, Des Monomers Polym. 3 (2000) 399-419. DOI:10.1163/156855500750206357 |
| [28] |
H.H. Moretto, M. Schulze, G. Wagner, "Silicones" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Wein-heim (2005). |
| [29] |
K.J. Shea, D.A. Loy, O. Webster, J. Am. Chem. Soc. 114 (1992) 6700-6710. DOI:10.1021/ja00043a014 |
| [30] |
Y. Liu, I. Imae, A. Makishima, Y. Kawakami, Sci. Technol. Adv. Mater. 4 (2003) 27-34. DOI:10.1016/S1468-6996(03)00021-4 |
| [31] |
U. Lauter, S.W. Kantor, K. Schmidt-Rohr, W.J. MacKnight, Macromolecules 32 (1999) 3426-3431. DOI:10.1021/ma981292f |
| [32] |
K. Nagaoka, H. Naruse, I. Shinohara, M. Watanabe, J. Polym. Sci. Part C:Polym. Lett. 22 (1984) 659-663. DOI:10.1002/pol.1984.130221205 |
| [33] |
G. Yi, J.S. Bradshaw, B.E. Rossiter, et al., J. Org. Chem. 58 (1993) 2561-2565. DOI:10.1021/jo00061a035 |
| [34] |
Y. Kawakami, I. Imae, ACS Symp. Ser. 838 (2003) 61-71. |
| [35] |
C. Cheng, A. Watts, M.A. Hillmyer, J.F. Hartwig, Angew. Chem. Int. Ed. 55 (2016) 11872-11876. DOI:10.1002/anie.201606282 |
| [36] |
Y. Li, Y. Kawakami, Macromolecules 32 (1999) 8768-8773. DOI:10.1021/ma991312t |
| [37] |
Y. Li, Y. Kawakami, Macromolecules 32 (1999) 6871-6873. DOI:10.1021/ma990511+ |
| [38] |
Y. Li, Y. Kawakami, Macromolecules 32 (1999) 3540-3542. DOI:10.1021/ma9819743 |
| [39] |
J. Cella, S. Rubinsztajn, Macromolecules 41 (2008) 6965-6971. DOI:10.1021/ma800833c |
| [40] |
J.J. Bozell, G.R. Petersen, Green Chem. 12 (2010) 539-554. DOI:10.1039/b922014c |
| [41] |
S. Vijjamarri, S. Streed, E.M. Serum, M.P. Sibi, G. Du, ACS Sustain. Chem. Eng. 6 (2018) 2491-2497. DOI:10.1021/acssuschemeng.7b03932 |
| [42] |
T. Kawakita, H.S. Oh, J.Y. Moon, Y. Liu, I. Imae, Polym. Int. 50 (2001) 1346-1351. DOI:10.1002/pi.784 |
| [43] |
R. Zhang, J.E. Mark, A.R. Pinhas, Macromolecules 33 (2000) 3508-3510. DOI:10.1021/ma000054t |
| [44] |
M. Oishi, J.Y. Moon, W. Janvikul, Y. Kawakami, Polym. Int. 50 (2001) 135-143. DOI:10.1002/1097-0126(200101)50:1<135::AID-PI600>3.0.CO;2-6 |
| [45] |
Y. Li, M. Seino, Y. Kawakami, Macromolecules 33 (2000) 5311-5314. DOI:10.1021/ma992143f |
| [46] |
S. Vijjamarri, V.K. Chidara, G. Du, ACS Omega 2 (2017) 582-591. DOI:10.1021/acsomega.6b00538 |
| [47] |
S. Vijjamarri, M. Hull, E. Kolodka, G. Du, ChemSusChem 11 (2018) 2881-2888. DOI:10.1002/cssc.201801123 |
| [48] |
X.Y. Zhai, S.B. Hu, L. Shi, Y.G. Zhou, Organometallics 37 (2018) 2342-2347. DOI:10.1021/acs.organomet.8b00316 |
| [49] |
H. Mutlu, M.A.R. Meier, Eur. J. Lipid Sci. Technol. 112 (2010) 10-30. DOI:10.1002/ejlt.200900138 |
| [50] |
K. Fujita, R. Kawahara, T. Aikawa, R. Yamaguchi, Angew. Chem. Int. Ed. 54 (2015) 9057-9060. DOI:10.1002/anie.201502194 |