 2020, Vol. 31
2020, Vol. 31 


b State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
Boron-containing π-electron systems, which integrate sp2-hybridized carbon skeleton and boron atoms, have drawn much attention in both synthetic chemistry and material science [1-6]. Till now, many B-containing π-systems have been developed for various optical and electronic applications, such as emissive materials in organic light-emitting devices (OLEDs), electron acceptors in organic solar cells (OSCs), semiconductors in organic field-effect transistors (OFETs), and electrode materials in lithium batteries, etc. [7-15]. In design of B-containing π-systems, the substituents on boron atoms are very important because they not only impact on the stability of the compounds but also significantly modulate the electronic structures and properties [16-24]. For example, the bulky aryl groups are usually incorpo-rated into tricoordinate B-containing compounds to sterically protect the boron atom due to its intrinsic instability towards oxygen and moisture. The substituents of tetracoordinate B-containing compounds can tune their light-absorption and fluorescence properties as well as electrochemical properties. Therefore, the development of new substituents on boron atom and investigation of their effects on π-systems remain crucial for organoboron chemistry and materials.
π-Systems containing boron-nitrogen coordination bond (B←N) as a typical kind of tetracoordinate organoboranes have received particular attention, because they have shown many intriguing properties, such as photo/thermo-chromism, thermally activated delayed fluorescence (TADF), and electron-transporting behavior [25-29]. Notably, B←N unit has been used to design n-type organic and polymer semiconductors for organic electronic applications [30-33]. For instance, twoB←N-containing azaacenes with the fluorine atoms on boron atoms (1a and 1b in Scheme 1) have been developed, which exhibited low-lying LUMO/HOMO energy levels (ELUMO/EHOMO) and excellent electron-transporting performance in OFETs [34].

|
Download:
|
| Scheme 1. Synthesis of 2a and 2b. | |
In this communication, we report that nucleophilic substitution reaction could proceed on 1a and 1b, producing two newB←N-containing azaacenes (2a and 2b) with the propynyl groups on boron atoms. They are good candidates to investigate the effects of the propynyl groups on π-systems. The theoretical and experi-mental results demonstrate that replacing fluorine atoms by propynyl groups significantly enhances the electronic energy levels, especially HOMO levels, and leads to some attractive optoelectronic properties, such as the near-infrared (NIR) light-absorption and fluorescence properties, as well as multiple reversible redox behaviors. The details are presented herein.
Scheme 1 shows the synthesis of 2a and 2b, through one-step Grignard reaction with 1-propynylmagnesium bromide. The nucleophilic substitution reactions proceeded completely on 1a and 1b, producing 2a and 2b in the yields of over 70% after purification by silica gel column chromatography [35]. The chemical structures of 2a and 2b are unambiguously characterized by 1H and 13C NMR spectra and elemental analysis. 2a and 2b are easily soluble in common organic solvents (> 20 mg/mL), such as chloroform, tetrahydrofuran and toluene, suggesting that the propynyl substituents can enhance the solubility of organoboron compounds. Thermogravimetric analysis (TGA) was used to characterize the thermal stability of 2a and 2b under nitrogen atmosphere. Upon increasing the temperature, an obvious weight loss of 8% at 273 ℃ was observed for 2a (Fig. S1 in Supporting information). Similarly, 2b exhibits a weight loss of 5% at 240 ℃. As calculated, the weight losses of 2a and 2b are ascribed to the release of two propynyl groups (theoretical values: 8.7% for 2a and 5.4% for 2b).
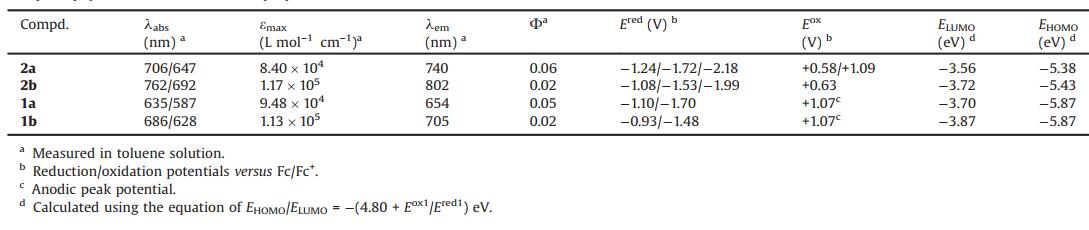
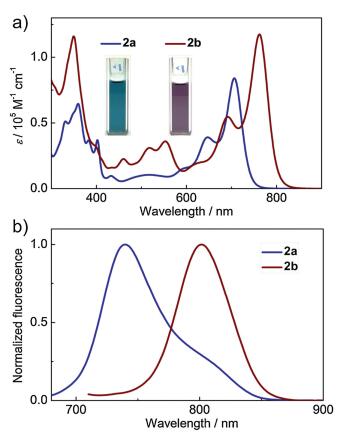
B←N-containing azaacenes with the propynyl groups display the interesting photophysical properties. The detailed data of 2a and 2b as well as 1a and 1b are summarized in Table 1. The solutions of 2a and 2b in toluene are deep blue and purple, respectively. As shown in Fig. 1a, the characteristic absorption bands in 300–800 nm are observed in their UV–vis spectra. While 2a shows the maximum absorption peaks (λabs) at 706/647 nm, 2b exhibits the λabs at 762/692 nm. The molar absorption coefficient is 8.40 104 L mol–1 cm–1 for 2a and 1.17 105 L mol–1 cm–1 for 2b, respectively. The redshifted absorption by ca. 50 nm from 2a to 2b and the higher light-absorption ability of 2b are attributed to the extended π-conjugation of 2b. These NIR absorption properties are rarely observed in organoboron compounds, which are mostly based on 4, 4'-difluoro-4-bora-3a, 4a-diaza-s-indacene (BODIPY) [36, 37]. Moreover, in comparison to 1a and 1b, 2a and 2b show the red-shifted absorption spectra by as large as ca. 70 nm, respec-tively, indicating that replacing fluorine atoms by propynyl groups on boron atoms greatly impacts on the absorption properties of B-containing conjugated molecules.
|
|
Table 1 The photophysical and electrochemical properties of 2a and 2b as well as 1a and 1b. |
{kind=link}
The fluorescence spectra of 2a and 2b exhibit the main emission bands in the NIR region with the peak maxima (λem) at 740 nm for 2a and 802 nm for 2b (Fig. 1b), respectively, although their fluorescence quantum yields (Φ) are moderate. The fluorescence peaks of 2a and 2b are significantly red-shifted by ca. 90 nm comparing with that of 1a and 1b. In addition, negligible solvent effects are shown in the fluorescence spectra of 2a and 2b as well as their absorption spectra (Figs. S3, S5 and S6 in Supporting information).

|
Download:
|
| Fig. 1. (a) UV–vis absorption spectra and (b) fluorescence spectra of 2a and 2b in toluene. Insets are the photographs of the two solutions. | |
{kind=link}
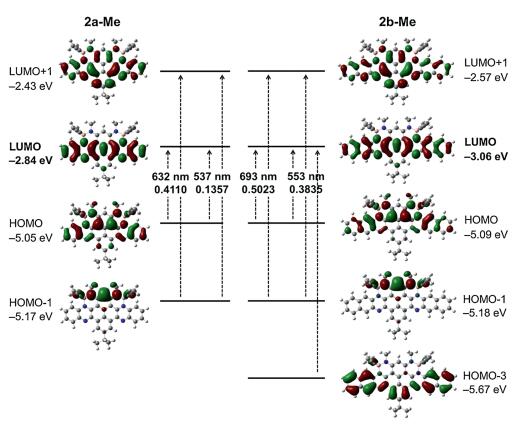
To illustrate the electronic effects of the propynyl groups, we performed density functional theory (DFT) calculations at the B3LYP/6-31 G(d, p) level of theory. The model compounds 2a-Me and 2b-Me with methyl groups were adopted for simplified calculations. The optimized structures of 2a-Me and 2b-Me both display the fully planar frameworks with four pendant propynyl groups (Fig. S7 in Supporting information). While the LUMOs of 2a-Me and 2b-Me are localized on the azaacene skeletons, the HOMOs are mainly distributed on the electron-rich alkylamine-linked phenyl rings and the peripheral rings of azaacene skeletons (Fig. 2). These LUMOs and HOMOs are quite similar to that of 1a-Me and 1b-Me, respectively, which have been reported in the previous work. It indicates that replacing fluorine atoms by propynyl groups has few effects on the molecular orbital distributions. Notably, in comparison to 1a-Me and 1b-Me, 2a-Me and 2b-Me exhibit the enhanced LUMO and HOMO energy levels, respectively, especially HOMO levels, thus leading to the narrow HOMO-LUMO bandgaps. This is ascribed to the weak electron-accepting character of the propynyl group, which significantly impacts on the HOMO levels of organoboron compounds.

|
Download:
|
| Fig. 2. Selected Kohn-Sham molecular orbitals and energy diagrams, as well as excitation energies and oscillator strengths of 2a and 2b calculated by DFT and TDDFT at the B3LYP/6-31 G(d,p) level, respectively. | |
{kind=link}
Time-dependent DFT calculations were conducted to illustrate the absorption properties of 2a and 2b. As shown in Fig. 2, the absorption bands at 706 and 647 nm of 2a are both assignable to the HOMO→LUMO and HOMO 1→LUMO+1 transitions. For 2b, the absorption bands at 762 nm and 692 nm are assigned to the HOMO→LUMO and HOMO 1→LUMO+1 transitions and the HOMO→LUMO, HOMO 1→LUMO+1 and HOMO 3→LUMO tran-sitions (Figs. S8 and S9 in Supporting information). The absorption spectra of 2a and 2b are well reproduced by the TD-DFT calculations, which all involve the HOMO→LUMO transitions. Namely, the NIR absorption properties of B←N-containing azaacenes with the propynyl groups are closely related to the narrowed HOMO-LUMO bandgaps. These calculations demon-strate that the electronic energy levels and absorption properties of organoboron compounds can be tuned through the incorporation of the propynyl groups on boron atoms.
To experimentally gain insight into the electronic properties, we measured the cyclic voltammetry of B←N-containing azaa-cenes 2a and 2b in CH2Cl2. Both of them show three reversible reduction processes, accompanying with two reversible oxidation processes for 2a and one reversible oxidation process for 2b. In contrast, 1a and 1b with fluorine atoms display the reversible reduction processes and the irreversible oxidation processes. It is suggested that the propynyl groups in 2a and 2b can stabilize not only the generated radical anion intermediates but also the resulted radical cation intermediates. The first half-wave reduction and oxidation potentials (Ered 1/Eox 1 vs. Fc/Fc+) are -1.24/+0.58 V for 2a and -1.08/+0.63 V for 2b. According to the equation of ELUMO/ EHOMO = -(4.80 + Ered 1/Eox 1), the experimental ELUMO and EHOMO of 2a and 2b are calculated to be -3.56/ -5.38 eV and -3.72/ -5.43 eV, respectively. The ELUMO and EHOMO of 2a are higher than that of 1a by 0.14 and 0.49 eV, respectively. Similarly, The ELUMO and EHOMO of 2b are enhanced by 0.15 eV and 0.44 eV from that of 1b. These changes are fully consistent with the theoretical calculation results. Thus, replacing fluorine atoms by propynyl groups plays an important role in tuning the electronic energy levels, especially enhancing the HOMO levels (Fig. 3).

|
Download:
|
| Fig. 3. Cyclic voltammograms of 2a and 2b in CH2Cl2 (0.4 mmol/L). Fc = ferrocene. | |
{kind=link}
In summary, we synthesized two new B←N-containing azaacenes with the propynyl groups on boron atoms through one-step Grignard reaction. The replacement of fluorine atoms by propynyl groups greatly impacts on the electronic energy levels, especially enhancing the HOMO levels, thus leading to the narrowed HOMO-LUMO bandgaps. These B←N-containing azaa-cenes with the propynyl groups exhibit the NIR light-absorption and fluorescence properties, as well as multiple reversible redox behaviors, which are significantly different from the analogs with fluorine atoms. This study thus provides a functional substituent of boron atom, which may lead to new organoboron materials with fascinating properties.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21822507, 21625403, 21761132020), National Key Research and Development Program of China (No. 2018YFE0100600), founded by MOST, Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2017265).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j. cclet.2019.11.018.
| [1] |
M. Hirai, N. Tanaka, M. Sakai, S. Yamaguchi, Chem. Rev. 119 (2019) 8291-8331. DOI:10.1021/acs.chemrev.8b00637 |
| [2] |
D. Frath, J. Massue, G. Ulrich, R. Ziessel, Angew. Chem. Int. Ed. 53 (2014) 2290-2310. DOI:10.1002/anie.201305554 |
| [3] |
A. Lorbach, A. Hübner, M. Wagner, Dalton Trans. 41 (2012) 6048-6063. DOI:10.1039/c2dt30118k |
| [4] |
S.K. Mellerupab, S. Wang, Chem. Soc. Rev. 48 (2019) 3537-3549. DOI:10.1039/C9CS00153K |
| [5] |
Y. Ren, F. Jäkle, Dalton Trans. 45 (2016) 13996-14007. DOI:10.1039/C6DT02756C |
| [6] |
P.G. Campbell, A.J.V. Marwitz, S.Y. Liu, Angew. Chem. Int. Ed. 51 (2012) 6074-6092. DOI:10.1002/anie.201200063 |
| [7] |
D. Li, H. Zhang, Y. Wang, Chem. Soc. Rev. 42 (2013) 8416-8433. DOI:10.1039/c3cs60170f |
| [8] |
Y.L. Rao, S. Wang, Inorg. Chem. 50 (2011) 12263-12274. DOI:10.1021/ic200658v |
| [9] |
Z.D. Zhuang, Z.H. Sun, Z.F. Yao, Q.R. Chen, Angew. Chem. Int. Ed. 58 (2019) 10708-10712. DOI:10.1002/anie.201905601 |
| [10] |
X.Y. Wang, F.D. Zhuang, R.B. Wang, X.C. Wang, et al., J. Am. Chem. Soc. 136 (2014) 3764-3767. |
| [11] |
W.B. Liu, M. Li, L. Fang, C.F. Chen, Chin. Chem. Lett. 29 (2018) 40-46. DOI:10.1016/j.cclet.2017.08.039 |
| [12] |
W. Zhang, G. Li, L. Xu, Y. Zhuo, et al., Chem. Sci. 9 (2018) 4444-4450. DOI:10.1039/C8SC00688A |
| [13] |
L. Wang, K.Q. Ye, H.Y. Zhang, Chin. Chem. Lett. 27 (2016) 1367-1375. DOI:10.1016/j.cclet.2016.06.049 |
| [14] |
C. Dou, J. Liu, L. Wang, Sci. China Chem. 60 (2017) 450-459. DOI:10.1007/s11426-016-0503-x |
| [15] |
C. Duan, G. Zango, M.G. Iglesias, F.J.M. Colberts, Angew. Chem. Int. Ed. 56 (2017) 148-152. DOI:10.1002/anie.201608644 |
| [16] |
L. Ji, S. Griesbeck, T.B. Marder, Chem. Sci. 8 (2017) 846-863. DOI:10.1039/C6SC04245G |
| [17] |
S. Yamaguchi, T. Shirasaka, S. Akiyama, K. Tamao, J. Am. Chem. Soc. 124 (2002) 8816-8817. DOI:10.1021/ja026689k |
| [18] |
L.G. Mercier, W.E. Piers, M. Parvez, Angew. Chem. Int. Ed. 48 (2009) 6108-6111. DOI:10.1002/anie.200902803 |
| [19] |
A.Jr Caruso, M.A. Siegler, J.D Tovar, Angew. Chem. Int. Ed. 49 (2010) 4213-4217. DOI:10.1002/anie.201000411 |
| [20] |
T. Wang, C. Dou, J. Liu, L. Wang, Chem. -Eur. J. 24 (2018) 13043-13048. DOI:10.1002/chem.201802496 |
| [21] |
M. Grandl, B. Rudolf, Y. Sun, D.F. Bechtel, et al., Organometallics 36 (2017) 2527-2535. DOI:10.1021/acs.organomet.6b00916 |
| [22] |
M. Grandl, Y. Sun, F. Pammer, Org. Chem. Front. 5 (2018) 336-352. DOI:10.1039/C7QO00876G |
| [23] |
D.L. Crossley, I.A. Cade, E.R. Clark, A. Escande, et al., Chem. Sci. 6 (2015) 5144-5151. DOI:10.1039/C5SC01800E |
| [24] |
X. Shao, C. Dou, J. Liu, L. Wang, Sci. China Chem. 62 (2019) 1387-1392. DOI:10.1007/s11426-019-9518-7 |
| [25] |
A. Wakamiya, T. Taniguchi, S. Yamaguchi, Angew. Chem. Int. Ed. 45 (2006) 3170-3173. DOI:10.1002/anie.200504391 |
| [26] |
Y.L. Rao, H. Amarne, S.B. Zhao, T.M. McCormick, et al., J. Am. Chem. Soc. 130 (2008) 12898-12900. DOI:10.1021/ja8052046 |
| [27] |
K. Liu, R.A. Lalancette, F. Jäkle, J. Am. Chem. Soc. 139 (2017) 18170-18173. DOI:10.1021/jacs.7b11062 |
| [28] |
K. Matsuo, T. Yasuda, Chem. Commun. (Camb.) 53 (2017) 8723-8726. DOI:10.1039/C7CC04875K |
| [29] |
Y.J. Shiu, Y.C. Cheng, W.L. Tsai, C.C. Wu, et al., Angew. Chem. Int. Ed. 55 (2016) 3017-3021. DOI:10.1002/anie.201509231 |
| [30] |
C. Dou, Z. Ding, Z. Zhang, Z. Xie, et al., Angew. Chem. Int. Ed. 54 (2015) 3648-3652. DOI:10.1002/anie.201411973 |
| [31] |
R. Zhao, C. Dou, J. Liu, L. Wang, et al., Chin. J. Polym. Sci. 35 (2017) 198-206. DOI:10.1007/s10118-017-1878-9 |
| [32] |
X. Long, C. Dou, J. Liu, L. Wang, Chin. Chem. Lett. 29 (2018) 1343-1346. DOI:10.1016/j.cclet.2018.01.052 |
| [33] |
Y. Min, C. Dou, H. Tian, J. Liu, et al., Chem. Commun. (Camb.) 55 (2019) 3638-3641. DOI:10.1039/C9CC00769E |
| [34] |
Y. Min, C. Dou, H. Tian, Y. Geng, et al., Angew. Chem. Int. Ed. 57 (2018) 2000-2004. DOI:10.1002/anie.201712986 |
| [35] |
L.J. Patalag, P.G. Jones, D.B. Werz, Angew. Chem. Int. Ed. 55 (2016) 13340-13344. DOI:10.1002/anie.201606883 |
| [36] |
Y. Ni, S. Lee, M. Son, N. Aratani, et al., Angew. Chem. Int. Ed. 55 (2016) 2815-2819. DOI:10.1002/anie.201511151 |
| [37] |
G.M. Fischer, E. Daltrozzo, A. Zumbusch, Angew. Chem. Int. Ed. 50 (2011) 1406-1409. DOI:10.1002/anie.201004829 |