 2020, Vol. 31
2020, Vol. 31 


Hepatocellular carcinoma (HCC) is one of the most common malignancies [1] and the survival rate of HCC patients is very poor, because most of the patients are diagnosed at the advanced stage, which is not suitable for hepatic transplantation, surgical resection and eradicative treatment [2]. Kinase inhibitor sorafenib (SRF) is the most widely used drug for advanced hepatocellular carcinoma, improving overall survival by inhibiting HCC cells proliferation and angiogenesis. Yet the therapeutic effects are seriously limited due to drug resistance [3]. Recent studies found several mechanisms are involved in the resistance, such as crosstalk between VEGFR/ MAPK and other signal pathways, including PI3K/Akt and JAK-STAT [4]. As a result, drug resistance can be overcome by targeting relative pathways.
MicroRNA-122 (miR122) is a liver-specific microRNA and considered as tumor suppressor microRNA which inhibits migra-tion, invasion, epithelial-mesenchymal transition (EMT), angio-genesis and the ability to form tumors [5]. Importantly, it is reported that restoration of miR122 sensitizes HCC cells to therapeutic drug [6]. Further study found that miR122 sensitizes hepatocellular carcinoma cells to SRF by targeting IGF1R to regulate MAPK pathway [7]. Thus, combination of SRF and miR122 has synergistic effects against HCC cells. However, a safe and efficient strategy to co-deliver both miR122 and SRF to disease sites is needed.
For decades, nanoparticle-based drug delivery systems (NDDSs) have attracted much attention for its unique property. Encapsula-tion of therapeutic molecules (e.g., RNAi, chemotherapy) can enhance cargo's solubility [8], prolong blood circulation time, accelerate target sites cellular uptake via the enhanced permeation and retention (EPR) effect [9], and reduce side effects on normal tissues. Passively targeted nanoparticles, only relying on EPR effect, are not sufficient to target tumor [10]. As a complementary strategy, actively targeted nanoparticles were developed. Actively targeted nanoparticles, which are modified with ligands on the surface, improve the cellular internalization of target cells [11] by receptor-mediated endocytosis. PEG segments are also used to improve delivery efficiency as they prolong circulation time of nanoparticles by providing "stealth" properties [12].
Co-delivery of gene and drug via a single delivery system offers several advantages over separate carriers. Encapsulation of two therapeutic agents into a single system endows the same pharmacokinetics and allows tumor cells to internalize both agents at a specific dose, which ensures that gene and drug work together to enhance anti-tumor effect [13]. Polymers are one of the main materials for constructing co-delivery system. However, most of the block polymers are synthesized via metal-based catalysts, as the residual catalysts may cause cytotoxicity and environmental pollution [14]. Relatively speaking, enzymatic catalysts show great potential because they are not only nontoxic and environmentally friendly, but also show higher catalytic activity [15].
Herein, we report a type of enzymatic synthesis block copolymer, galactose-poly(ethylene glycol)-poly(ω-pentadecalactone-co-N-methyldiethyleneamine-co-sebacic acid) (Gal-PEG-PPMS), which can self-assemble into functional positively charged micelles in sodium acetate buffer at pH 5.2 to bind miR122. Meanwhile, PPMS segments are used as core to encapsulate SRF through hydrophobic effect. Furthermore, galactose (Gal) modification on PEG-PPMS micelles could enhance the targeting to asialoglycoprotein receptor (ASGPR) on hepatocytes [16].
The synthesis of Gal-PEG-PPMS polymers was carried out as Scheme 1. Firstly, PPMS-COOH polymer was synthesized via CALB-mediated copolymerization of ω-pentadecalactone (PDL), N-methyldiethanolamine (MDEA) and sebacic acid (SA). The molar ratio of PDL:MDEA:SA is 15:85:85. The GPC analysis showed polymers had molecule weight (Mw) value of 28, 500 Da with Mw/Mn of 3.5. Meanwhile, Gal-PEG5k-NH2 was synthe-sized by conjugating HOOC-PEG5k-NH2 to 4-aminophenyl β-D-galactopyranoside.

|
Download:
|
| Scheme 1. Synthesis of Gal-PEG5k-PPMS polymers. | |
To maximum the galactose conjunction, the number moles of 4-aminophenyl β-D-galactopyranoside was adjusted to be 3-fold as moles of HOOC-PEG5k-NH2. The structure was characterized by 1H NMR spectroscopy and the detailed assignments were showed (Fig. S1A in Supporting information). The amphiphilic polymers, Gal-PEG-PPMS and PEG-PPMS, were synthesized by carbodiimide chemistry, followed by dialysis against DMSO and the structure was characterized by 1H NMR spectroscopy (Figs. S1B and S1C in Supporting information).

|
Download:
|
| Fig. 1. (A) Size distribution of Gal-PEG-PPMS blank micelles. (B) TEM image of Gal-PEG-PPMS blank micelles (Scale bar: 50 nm). (C) Size variations of PEG-PPMS and Gal-PEG-PPMS micelles in PBS solution containing 10% FBS. (D) Relative fluorescence intensity of gel-red incubated with different samples. Data are expressed as mean±SD (n = 3). | |
Gal-PEG5k-NH2 polymer: 1H NMR (DMSO-d6):δ 3.52 (s), 6.97 (br.), 7.49 (br.), 9.81 (s); Gal-PEG5k-PPMS polymer: 1H NMR (DMSO-d6):δ 2.51 (t), 2.62 (t), 3.52 (s), 4.06 (t), 6.97 (br.), 7.48 (br.), 9.81 (s).
The amphiphilic polymers can self-assemble into micelles after dialysis at sodium acetate buffer (25 mmol/L, pH 5.2). The relative characterizations of micelles were measured and shown in Table 1. Micelles with a low polydispersity index (PDI) ranging from 0.13 to 0.15 proved its uniform size distribution (Table 1). The average size of PEG-PPMS micelles is 165.1 7.3 nm, and the size of Gal-PEG-PPMS micelles is slightly larger (195.1 8.3 nm), which are beneficial to reducing reticuloendothelial system (RES) clearance and enhancing EPR effect. The size distribution and transmission electron microscope (TEM) images of Gal-PEG-PPMS micelles were also shown (Figs. 1A and B).
|
|
Table 1 Characterization of the co-delivery micelles in ddH2O. |

{kind=link}
{kind=link}
Gal-PEG-PPMS micelles have spherical morphology with uniform size distribution and were dispersed well. The size of micelles in TEM image was slightly smaller than that in Table 1 because when measured by DLS in ddH2O, the PEG layer formed hydration shell on the surface of micelles. The surface charge of miR122-loaded micelles (range of 6.8–7.3 mV) was slightly positive and lower than that of blank micelles (range 16.9–18.2 mV) (Fig. S2 in Supporting information) due to negatively charged miR122 mimic. It is well-known that nanoparticles with neutral charge (ranging from-10 mV to+10 mV) on the surface can extend blood circulation by lessening binding with serum protein [17]. The drug loading (DL) and entrapment efficiency (EE) were measured by HPLC. The EE for PEG-PPMS and Gal-PEG-PPMS micelles was 71.9% and 68.8%, and the DL capacity was 3.43% and 3.28%, respectively (Table 1), indicating their excellent drug loading capability.
Besides the capability to load drug and gene, the stability of nanoparticles in serum solution is important for preventing drug release in the blood stream. The colloidal stability of micelles was evaluated. Typically, PEG-PPMS and Gal-PEG-PPMS micelles were incubated with PBS solution (10 mmol/L, pH 7.4) containing 10% FBS for 5 days and the hydrodynamic diameters of micelle was measured at various intervals by DLS. Obviously, there was no significant size change after incubation during experiment time (Fig. 1C), due to the existence of PEG segments which avoid interaction with serum protein, indicating that micelles were stable sufficiently and suitable for in vivo application. Sufficient condensation of RNA/DNA into micelles is important for effective gene delivery. The condensation ability of micelles was explored by a gel-red exclusion assay. The relative fluorescence intensity of solutions incubation with PEG-PPMS and Gal-PEG-PPMS micelles and LPF2k is 38.9% and 34.8%, 2.81%, respectively (Fig. 1D), indicating that micelles have high RNA condensing capability for further study.
Burst release of drug is a common but serious problem of drug-loaded micelles. Most micelles release most of drug in initial 24 h, which is adverse for drug delivery. The in vitro release behavior of SRF was tested by a dialysis method. The release profile was showed in Fig. S3 (Supporting information). At initial 7 days, drug was continually released from micelles at a similar speed. At the end of 7 days, 51.38% and 47.97% of SRF were released from PEG-PPMS and Gal-PEG-PPMS micelles, respectively. For the last 3 days, less than 10% of SRF was released from both micelles. The slow release speed of SRF from micelles is attributed to the thick PEG5k shell. As previously reported, the diffusion of hydrophobic SRF across thick PEG5k shell from PPMS core to outer medium was slow [18]. The result indicated that the micelles had the ability to achieve long-term sustained drug release.
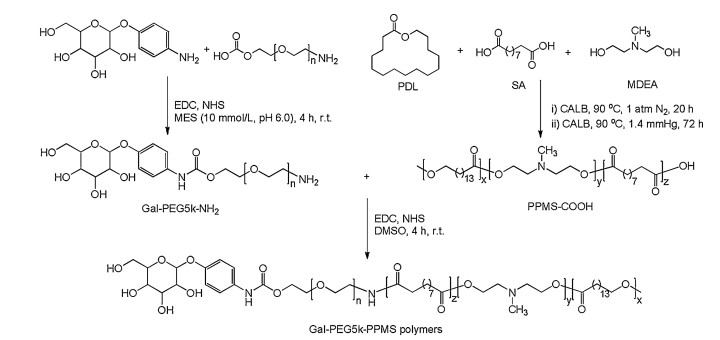
Biocompatibility of drug vector affects its application greatly. Vector with high cytotoxicity has restricted the application of many carriers. The cytotoxicity of blank micelles against HepG2 cells was evaluated by MTT assay. Generally, cell viabilities decreased with the increase of polymer concentration from 25 μg/mL to 400 μg/mL. Importantly, the average cell viabilities of PEG-PPMS and Gal-PEG-PPMS micelles were over 80% (81.3% and 81.6%, respectively) even at a polymer concentration of 400 μg/mL (Fig. 2A). Obviously, polymer micelles showed minimal cytotoxici-ty to cells. The hemolytic activity of blank micelles was investigated. The result showed the hemolytic rate was low at all polymer concentrations (Fig. 2B). Even when the polymer concentration was as high as 400 μg/mL, the hemolytic rate after incubated with PEG-PPMS and Gal-PEG-PPMS micelles (4.20% and 4.24%, respectively) was less than 5%, confirming their excellent biocompatibility.

|
Download:
|
| Fig. 2. (A) Viabilities of HepG2 cells after incubation with blank micelles for 48 h.(B) Hemolytic rate of RBCs after incubation with blank micelles for 2 h. Data are given as mean±SD (n = 3). | |
{kind=link}
To verify the target ability of Gal-micelles, we explored cell uptake activity and distribution of HepG2 cells which expresses a great amount of AGSPR on the cell surface (Fig. 3A). Obviously, the MFI of cells is time-dependent and the maximum value of MFI is at 2 h. Compared to PEG-PPMS micelles, the mean fluorescence intensity of HepG2 cells incubated with Gal-PEG-PPMS micelles was higher. For example, MFI of cells incubated with PEG-PPMS and Gal-PEG-PPMS micelles for 4 h was 672 and 864.5, respectively, in which mean MFI of cells incubated with Gal-micelles was 1.3-fold higher, demonstrating the higher uptake efficiency of Gal-micelles. CLSM was utilized to visualize cellular transportation of micelles on HepG2 cells. Stronger intracellular fluorescence intensity of C6 was observed in cells treated with Gal-PEG-PPMS micelles relative to PEG-PPMS micelles after 4 h incubation. These result illustrated that galactose residues modification on the surface of micelle can promote HepG2 cells uptake. Competitive inhibition assay was conducted to explore the role of ASGPR in the augment of cell uptake. When treated with 65 mmol/L free galactose before incubation, HepG2 cells incubated with Gal-PEG-PPMS micelles exhibited similar fluorescence intensity with cells incubated with PEG-PPMS micelles (Fig. 3B). The enhanced cell uptake is attributed to ASGPR-mediated endocytosis. If ASGPR on the surface is blocked by free galactose, the binding affinity between cells and Gal-micelles wound will decrease, which lead to similar MFI relative to cells treated with PEG-PPMS micelles. The CLSM images were in consistence with the result obtained by flow cytometry (Fig. 3C). Results above demonstrated the Gal-PEG-PPMS micelles as an efficient carrier for targeting liver cancer.

|
Download:
|
| Fig. 3. (A) MFI of HepG2 cells after incubation with C6-loaded micelles at different time points. (B) Competitive inhibition assay. Before incubating with C6-loaded micelles, HepG2 cells were pretreated with 65 mmol/L galactose. Data are given as mean±SD (n = 2). (C) CLSM images of HepG2 cells incubated with C6-loaded micelles (green). Hoechst 33342 (blue) was used to visualize nucleus. The scale bars are 10 μm and * represents P < 0.05. | |
{kind=link}
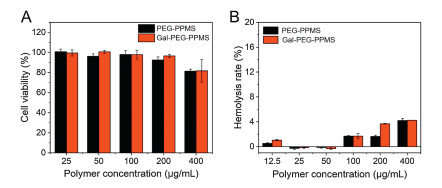
To testify the function of miR122 mimic after being loaded in micelles, the real-time PCR assay was carried out to measure miR122 level of HepG2 cells (Fig. 4A). After incubation with PEG-PPMS/miR122, PPMS-PEG-Gal/miR122 or LPF2k/ miR122, the relative miR122 levels of cells were 11.05, 17.25 and 53.83, respectively. The result showed that polymeric micelles did not affect the function of miR122 mimic and greatly promoted miR122 level in HepG2 cells. Importantly, due to the higher cellular uptake of Gal-micelles, cells treated with PPMS-PEG-Gal/miR122 micelles expressed higher miR122 level, further demonstrating the target-ing capability of Gal-PEG-PPMS micelles.

|
Download:
|
| Fig. 4. (A) miR122 level in HepG2 cells. (B) Wound closure percentage of scratch after transfected with miR122-loaded micelles. (C) Microscope images of scratch after various treatments. (D) Invasion percentage of cells after transfected with miR122-loaded micelles. (E) Optical images of cells after various treatments. The scale bar is 40 μm. Data are given as mean±SD (n = 3). ** represents P < 0.01, *** represents P < 0.001. | |
{kind=link}
To further investigate the bioactivity of miR122 mimic, the migration and invasion ability of HCC cells were explored on HCC cells. Because of the strong migration and invasion capability, SMMC7721 cells were investigated. For wound healing assay, cells in a 6-well plate were treated with micelles/miR122 (or negative control RNA) for 24 h and the migration behavior within another 24 h was explored. Great inhibition of cell migration capacity was observed after treated with miR122-loaded micelles, and cells incubated with micelles/NC did not show the same result (Figs. 4B and C). For example, the wound closure percentages of cells were 40.82% and 37.26% in PEG-PPMS/miR122 and Gal-PEG-PPMS/ miR122 micelles treated group, and 84.60%, 76.03% and 76.94% in control group, PEG-PPMS/NC and Gal-PEG-PPMS/NC group, respectively. The optical images of cells also exhibited similar result. Clearly, scratch was larger after incubation with miR122-loaded micelles, indicating the inhibition of migration capability.
Furthermore, the invasion capability of cells was explored on SMMC7721 cells by Transwell chamber. The percentage of invasive cells after transfected with miR122-loaded micelles decreased significantly, indicating the suppress of cell invasion capability (Figs. 4D and E). Collectively, Gal-PEG-PPMS micelles can keep the function of miR122 and then inhibit the migration and invasion ability of HCC cells by up-regulating miR122 level.
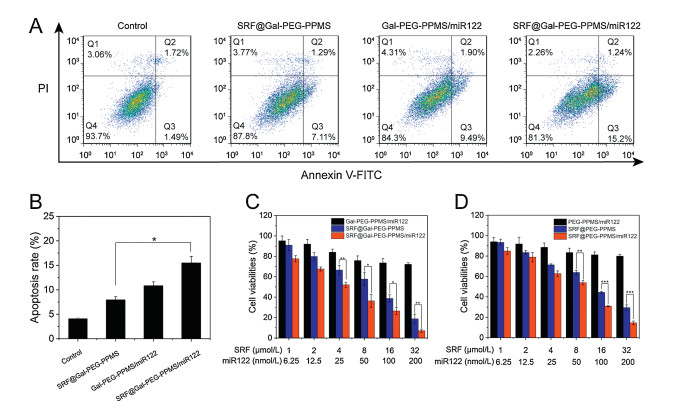
Up-regulation of miR122 level sensitizes HCC cells to SRF, so the synergistic effect of SRF and miR122 was further explored. As shown in Figs. 5A and B, higher cell apoptosis was observed in group treated with co-delivery micelles, suggesting enhanced ability of co-delivery micelles to induce cell apoptosis.

|
Download:
|
| Fig. 5. (A) Flow cytometry images of HepG2 cell after various treatments. (Q1: dead cells; Q2: late apoptotic cells; Q3: early apoptotic cells; Q4: live cells). (B) Quantitative analysis of cell apoptosis. (C) Cell viabilities of HepG2 cells after incubation with different Gal-PEG-PPMS micelles. (D) Cell viabilities of HepG2 cells after incubation with different PEG-PPMS micelles. Data are given as mean±SD (n = 3) | |
{kind=link}
Moreover, the synergistic effect was also evaluated by MTT assay (Figs. 5C and D). The cell viabilities decreased with the increase of drug concentration. The miR122-loaded micelles did not exhibit obvious inhibition on cell proliferation. Compared to SRF@Gal-PEG-PPMS micelles, SRF@Gal-PEG-PPMS/miR122 micelles resulted in lower cell viabilities, indicating the enhanced ability of co-delivery micelles to inhibit cell proliferation. Exactly, IC50 values for SRF@Gal-PEG-PPMS, SRF@Gal-PEG-PPMS/miR122 micelles were calculated to be 8.72 and 4.32 μg/mL, respectively. The similar experiment result appeared in the cells treated with PEG-PPMS micelles, whose IC50 values in SRF@PEG-PPMS and SRF@PEG-PPMS/miR122 group were 12.4 and 6.96 μg/mL respectively. The stronger inhibition effect observed was attributed to the addition of miR122. As reported previously, miR122 can sensitizes HCC cells to SRF by regulating MAPK pathways, which is associated with drug resistance [7]. Compared to single treatment, co-delivery of SRF and miR122 could overcome SRF resistance, thus showed more significant inhibition effect.
Furthermore, co-delivery of gene and drug using a single carrier is challenging because of their different physicochemical properties [19]. Nano-carrier for co-delivery is required to be non-toxic, stable and capable of efficiently condensing gene, as well as encapsulating drug. In this study, in order to co-deliver SRF and miR122, Gal-PEG-PPMS polymeric micelle system was developed. And such micelles were stable and had gradual sustained drug release profile of SRF and desirable gene transfection efficiency. Besides, Gal-micelles enhanced the uptake of HepG2 cells owing to ASGPR-mediated endocytosis. The polymer micelles could efficiently improve miR122 level and inhibit the migration and invasion capability of HCC cells. Importantly, synergistic effect on inducing cell apoptosis and inhibiting cell proliferation was observed. As a result, the Gal-PEG-PPMS micelles are promising nano-carriers for target delivery of therapeutic drug and gene to treat HCC.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (No. 51773231), the Natural Science Foundation of Guangdong Province (Nos. 2016A030313315, 2014A030312018), and the Project of Key Laboratory of Sensing Technology and Biomedical Instruments of Guangdong Province (No. 2011A060901013).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.030.
| [1] |
F. Bray, J. Ferlay, I. Soerjomataram, et al., CA Cancer J. Clin. 68 (2018) 394-424. DOI:10.3322/caac.21492 |
| [2] |
D. Lee, I.M.J. Xu, D.K.C. Chiu, et al., J. Clin. Invest. 127 (2017) 1856-1872. DOI:10.1172/JCI90253 |
| [3] |
R. Rudalska, D. Dauch, T. Longerich, et al., Nat. Med. 20 (2014) 1138-1146. DOI:10.1038/nm.3679 |
| [4] |
F. Feng, Q. Jiang, S. Cao, et al., Biochim. Biophys. Acta-Gen. Subj. 1862 (2018) 1017-1030. DOI:10.1016/j.bbagen.2018.01.011 |
| [5] |
S.H. Hsu, B. Wang, J. Kota, et al., J. Clin. Invest. 122 (2012) 2871-2883. DOI:10.1172/JCI63539 |
| [6] |
G. Lou, X. Song, F. Yang, et al., . Hematol. Oncol 8 (2015) 122. DOI:10.1186/s13045-015-0220-7 |
| [7] |
Y. Xu, J. Huang, L. Ma, et al., Cancer Lett. 371 (2016) 171-181. DOI:10.1016/j.canlet.2015.11.034 |
| [8] |
D. Rosenblum, N. Joshi, W. Tao, J.M. Karp, D Peer, Nat. Commun 9 (2018) 1410. DOI:10.1038/s41467-018-03705-y |
| [9] |
N. Kamaly, J.C. He, D.A. Ausiello, O.C. Farokhzad, Nat. Rev. Nephrol. 12 (2016) 738-753. DOI:10.1038/nrneph.2016.156 |
| [10] |
S. Wilhelm, A.J. Tavares, Q. Dai, et al., Nat. Rev. Mater. 1 (2016) 16014. DOI:10.1038/natrevmats.2016.14 |
| [11] |
D.B. Kirpotin, D.C. Drummond, Y. Shao, et al., Cancer Res. 66 (2006) 6732-6740. DOI:10.1158/0008-5472.CAN-05-4199 |
| [12] |
S. Schottler, G. Becker, S. Winzen, et al., Nat. Nanotechnol. 11 (2016) 372-377. DOI:10.1038/nnano.2015.330 |
| [13] |
P.Y. Teo, W. Cheng, J.L. Hedrick, Y.Y. Yang, Adv. Drug Deliv. Rev. 98 (2016) 41-63. DOI:10.1016/j.addr.2015.10.014 |
| [14] |
A. Corma, S. Iborra, A. Velty, Chem. Rev. 107 (2007) 2411-2502. DOI:10.1021/cr050989d |
| [15] |
Z.Z. Jiang, Biomacromolecules 11 (2010) 1089-1093. DOI:10.1021/bm1000586 |
| [16] |
Z. Ye, W.R. Wu, Y.F. Qin, et al., Adv. Funct. Mater 28 (2018) 1706600. DOI:10.1002/adfm.201706600 |
| [17] |
M.P. Leal, C. Caro, M.L. Garcia-Martin, Nanoscale 9 (2017) 8176-8184. DOI:10.1039/C7NR01607G |
| [18] |
X.F. Zhang, B. Liu, Z. Yang, et al., Colloids Surfaces B 115 (2014) 349-358. DOI:10.1016/j.colsurfb.2013.12.029 |
| [19] |
J.A. Kemp, M.S. Shim, C.Y. Heo, Y.J. Kwon, Adv. Drug Deliv. Rev. 98 (2016) 3-18. DOI:10.1016/j.addr.2015.10.019 |