 2020, Vol. 31
2020, Vol. 31 


b Department of Pancreatic Surgery, West China Hospital, Sichuan University, Chengdu 610041, China;
c College of Pharmacy, Southwest Minzu University, Chengdu 610041, China
Peptide drugs are increasingly being applied in clinic for different diseases therapy mainly by parenteral routes [1]. The oral administration of most peptides remained severely restricted by the physiological barriers of gastrointestinal tract (GI), such as poor permeability across the mucus and epithelium and rapid degradation [2]. The nanotechnology has opened up a new perspective to the oral delivery of peptides [3]. Numerous oral nanomedicines loaded with peptides have been reported. However, it is still challenging to overcome both the mucus and the epithelium barrier in the design of nanocarriers for efficient peptide delivery by the oral route [4]. The gastro intestinal mucus covers the epitheliums a layer of slippery secretion consisting of protein fibers, which can trap nanomedicines [5, 6]. Although lots of studies have been performed to overcome the mucus barrier, it should be noted that the physicochemical properties of nanoparticles for mucus permeation are significantly different from those for epithelial absorption [6]. The rigidity of nanoparticles was newly found to play a governing role in oral delivery of nanoparticles, especially for penetration in mucus and uptake by epithelial cells [5]. However, this novel strategy has not been applied for the development of oral peptide drugs. Here, we focus on an oral peptide formulation which can achieve enhanced bioavailability.
With rapid development in nanotechnology, many advantages of liposomes and nanoparticles for drug delivery have been recognized [7-9]. In those nanotechnology based drug delivery systems, PLGA nanoparticles was one of the most widely used nanocarriers with numerous advantages [10], such as high structural integrity, controlled release profiles, biodegradability and low toxicity. Whereas, there is evidence implying that the physicochemical properties of nanoparticles are often selfcontradictory, and they often fail to balance efficient cellular internalization and effective penetration [11, 12]. We have designed PLGA nanoparticles for oral peptide drug delivery [13], which could improve the pharmacokinetics in animals than free peptide. However, there still remain some unsatisfying disadvantages of polymer nanoparticles to be solved such as drug leakage [11], limited affinity to the intestinal epithelial cells [12, 14, 15]. To address these issues, modifying nanoparticles with other materials has emerged as an attracting strategy. Nanoparticles with intermediate rigidity (i.e., semi-elastic nanoparticles) were reported to have superior diffusion through various intestinal barriers by M. Yu and his coworkers [5]. The semi-elastic core-shell poly(lactic-co-glycolic acid) (PLGA)-lipid nanoparticles (LNPs) loaded with doxorubicin can improve the bioavailability compared to doxorubicin solution after oral administration. The lipid nanoparticles, combining the advantages of both liposomes and nanoparticles [16-19], have obtained tremendous attention to ameliorate these limitations [5, 19] and have been recently used as a common nanovesicle for oral administration, owing to the excellent drug loading rate, drug protection against GI degradation and sustained-release profile [18, 20, 21]. However, it remains unclear that whether the semi-elastic LNPs can enhance the oral bioavailability of peptide drugs.
Inspired by findings that elastic core-shell nanoparticles could achieve the rapid transport across the biological hydrogels and cellular barriers, core-shell nanoparticles were designed loaded small peptide as oral peptide delivery systems to enhance bioavailability than regular nanoparticles. We herein developed a semi-elastic core-shell LNPs for oral delivery of a peptide drug, which was designed with a new strategy. With Val-Leu-Pro-ValPro (VP5) as a model drug, the lipid nanoparticles contained a VP5/PLGA nanoparticle-core and lipid shell. VP5 is one of antihypertensive small peptides with in vitro inhibitory activity of angiotensin converting enzyme (ACE) (IC50 1.8 μmol/L) and in vivo antihypertensive effect [22]. However, its poor oral bioavailability largely limited the effectiveness and the active duration of treatment. PLGA, approved by FDA and EMA, is a well-known bio-degradable and biocompatible polymer [23, 24]. It has been used in encapsulating a variety of hydrophilic peptide drugs for effectively protection from degradation in the GI tract [13, 25-27]. Nevertheless, the oral bioavailability of PLGA nanoparticles still needs to be further enhanced, probably because they are too rigid to cross intestinal barriers with high efficiency. In this study, the semi-elastic LNPs were validated for the first time as the carriers of oral peptide drugs. The processing parameters of VP5 loaded LNPs (VP5- LNPs) were optimized to achieve high entrapment efficiency, and then the in vitro cellular uptake of VP5-LNPs was investigated. The in vivo pharmacokinetic profiles of VP5-LNPs finally demonstrated that the semi-elastic LNPs can successfully improve the oral bioavailability of VP5 compared to the rigid PLGA nanoparticles.
The elastic LNPs loaded VP5 were prepared by a two-step approach. Lipid film was formed and then hydrated with VP5-NPs suspension to obtain VP5-LNPs. Firstly, VP5/PLGA nanoparticles (NPs, VP5-NPs) were prepared by a double-emulsion (W1/O/W2) solvent evaporation method as previously reported [13, 28, 29]. The VP5-NPs were further coated by membrane hydration- homogeneity dispersion method to obtain the core-shell lipid nanoparticles (VP5-LNPs). In brief, 50 μL VP5 (10 mg/mL) deionized water solution was used to form the inner aqueous phase. The 1 mL organic acetonitrile phase containing PLGA was used as the organic phase, and then mixed with the inner aqueous phase by probe sonication in ice bath to form the primary W1/O emulsion. The primary emulsion was then added to the 4 mL external phase solution and further sonicated to obtain the final W1/O/W2 double emulsion. The organic phase in the ultimate emulsion was rapidly removed by evaporation under vacuum at 37 ℃ to get VP5-NPs [13]. At the same time, PC (lipid), Chol and mPEG2000- Cholmixture (ratio were: 4:1:0.25) were dissolved in 2 mL chloroform at a certain proportion. The organic solvents were subsequently removed using a rotary evaporator (R-201Shanghai Shen Shun Biological Technology Co., Ltd.) to produce a thin film of lipid at 37 ℃. The lipid film was hydrated with 4 mL prepared VP5- NPs for 1 h at 60 ℃ to obtain a suspension. VP5-LNPs were finally acquired followed by homogeneous of the above suspension by Ultrasonic cell crushing and isolating machine (VCX130, American systems on ICs) for 3 min in an ice bath. The coumarin-6 (Cou-6) loaded NPs or LNPs were constructed using the similar procedure but the addition of Cou-6 into the solvent when preparing of VP5-NPs. The unpackaged VP5 was separated from VP5-LNPs by ultra filtration (Millipore, 10, 000 Da) and evaluated by high performance liquid chromatography (HPLC, Waters Alliance 2695). The filtrate after centrifuging was used to determine the EE and drug loading (DL) of VP5-LNPs as previously described [13]. Analyses were performed in triplicate and the values were expressed as mean ± SD. EE% and DL% of VP5 were calculated by the following formula:

|
The mean particle size, size distribution and ζ potential were measured by Zetasizer (Zetasizer Nano-ZS 90, Malvern Instruments Ltd., Malvern, UK) at 25 ℃. The prepared VP5-LNPs were diluted with deionized water and submitted to size and ζ potential experiments in triplicate. All the data were presented as mean ± SD.
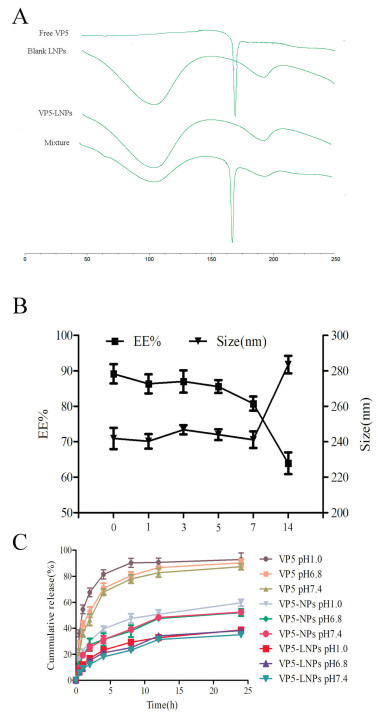
As was known, EE% and size are critical properties for nanocarriers, which affect stability, drug release profile and absorption of preparation at the nanoscale [30]. Therefore, different mole ratios of PC versus Chol (PC/Chol) and PLGA versus lipid bilayer materials (PLGA/LP materials) were investigated to establish the optimal preparation conditions for VP5-LNPs. Cholesterol could play a critical role in the flexibility and the arrangement of lipid membrane in the preparation process [31]. Too much or less Cholesterol both could affect the form of lipid nanoparticles. Impropriety content of Chol might cause the decreasing encapsulation of the drug. As seen in Fig. 1A, the entrapment content of VP5 first increased and then decreased with the decrease of cholesterol, while the particle size has changed slightly as the cholesterol content changed. When the PC was the fourth molar amount of cholesterol, LNPs could entrap more VP5 (57.87% ± 3.75%) with satisfied particle size (221.1 ± 2.3 nm). Besides, the proportion of polymer to lipid materials was thought to be another influential parameter affecting particle EE% and size of lipid nanoparticles [32, 33]. The EE and the size of particles were further optimized using different molar ratio of PLGA/LP materials. The drug content of VP5 ranged between 63.38% ± 3.89% and 89.88% ± 4.07% and the size of VP5-LNPs ranged between 232.2 ± 5.2 nm and 247.3 ± 3.8 nm depending on the ratios of PLGA/LP materials. Fig. 1B showed that the higher EE% and the better particle size of VP5-LNPs was gained at 1:1.5 ratio of PLGA/LP materials, which might result from the increased affinity between drug and the formulation with the increase of lipid materials proportion [34]. Overall, the optimal VP5-LNPs were successfully prepared with 4:1 molar ratio of PC/Chol and 1:1.5 ratio of PLGA/LP materials with an EE% of 89.88% ± 1.23% and DL% of 1.54% ± 0.25%. The average particle size of optimal VP5-LNPs was 247.3 ± 3.8 nm (Fig. 1C) with a narrow size distribution (PDI = 0.09 ± 0.02). The Zeta potential of VP5-LNPs was -6.57 ± 0.45 mV (Fig. 1D). The colloidal solution was observed as slightly blue opalescence with strong Tyndall effect (Fig. 1E). As shown in Fig. 1F, the shape of the nanoparticles was sphere, while the liposomes were nearly spherical with distinct lipid membrane structure because of the different preparation processes as reported. The size of LNPs was larger than that of nanoparticles almost 20 nm, and the shape of LNPs exhibited irregular oval with obvious shadow. The shadow is considered to be the kernel of the nanoparticles. Thus, we suggested that the core-shell lipid nanoparticles were formed successfully and the lipid bilayer was well coated on the nanoparticles according to the TEM image.

|
Download:
|
| Fig. 1. Optimization of VP5-LNPs. (A, B) Diameter and EE% of VP5-LNPs at various ratios of PC/Chol (A) and PLGA/LP materials (B), respectively. (C) Size distribution, (D) zeta potential, (E) appearance and Tyndall effect and (F) TEM images of optimal VP5-LNPs (n = 3). | |
To investigate the existing state of VP5 and verify the entrapment of VP5 into lipid nanoparticles, DSC analysis was performed. The physical state of VP5 loaded in VP5-LNPs was measured by DSC (200PC, Netzsch, Karlsruhe, Germany). Freezedried VP5-LNPs, blank LNPs, VP5 and the physical mixture of blank LNPs and free VP5 with the same mass ratio as those in VP5-LNPs were heated from 40 ℃ to 250 ℃ at a heating ramp of 20 ℃/min under nitrogen atmosphere at a flow rate of 50 mL/min. As was shown in Fig. 2A, the free VP5 presented a sharp endothermic peak at 167.0 ℃ probably caused by its degradation. However, VP5-LNPs performed no endothermic peak at 167.0 ℃ in DSC thermo gram, while there were only two wide endothermic peaks at 118.2 ℃ and 193.6 ℃ resulted from the glass transition temperature of the PLGA and PVA, respectively [35-37]. Interestingly, the physical mixture showed three endothermic peaks which attributed to the free VP5 and the materials. The result indicated that VP5 formulated in the lipid nanoparticles existed as an amorphous state or a solid solution in the polymeric matrix in the formulation process [13, 18, 30], which implied that the VP5 was successfully entrapped in VP5-LNPs.

|
Download:
|
| Fig. 2. Characterization of optimal VP5-LNPs. (A) DSC curves of free VP5, blank LNPs, VP5-LNPs and the physical mixture of VP5 and blank LNPs. (B) Change in size and EE% of VP5-LNPs over 14 days stored at 4 ℃. (C) Release profiles of free VP5, VP5-NPs and VP5-LNPs in phosphate buffers (pH 1.0, 6.8 and 7.4). | |
In addition, the core-shell VP5-LNPs displayed a good stability with no remarkable changes in EE% and particle sizes for at least 5 d (Fig. 2B). When stored over 1 week, a sharp drop of EE% and an enlargement of particle size of VP5-LNPs were found. In summary, VP5-LNPs were suggested to be stable for five days at 4 ℃ and be further investigated to develop a freeze-drying formulation.
It is well known that in vitro release can simulate the possible release in vivo. Therefore, the in vitro release profiles of VP5-LNPs by dynamic dialysis method [13, 18, 38] in different environment (pH 1.0, 6.8 and 7.4) were illustrated in Fig. 2C. Free VP5 solution, VP5-NPs, and VP5-LNPs were firstly dispersed in release media in dialysis bags (MWCO 3000) and then shook at 37 ℃ with a speed of 100 rpm. At given time interval (0.5, 1, 2, 4, 6, 8, 12 h), 1 mL buffer was removed and replaced with equal volume fresh release medium. As we can see, VP5 was released gradually from VP5-LNPs without remarkable burst release in different release medium. Within a 12 h period, almost all of free VP5 were released at pH 1.0 and 6.8, over half of the VP5 was released from VP5-NPs, while only less than 40% of VP5 was released from VP5-LNPs, which might lead to a potent and prolonged therapeutic efficacy of VP5-LNPs. The result implied that the amazing semi-elastic core-shell structure could efficiently protect the drug from gastric acid-like damage and also expected to achieve sustained release in the intestinal tract, which could result tunable and sustained drug release profiles [31]. However, the probable mechanism was needed to be evaluated in the future.
However, the possible transcelluar mechanism of core-shell LNPs is unclear. Hence, the cellular uptake study of LNPs was evaluated in Caco-2 cells and HT-29 cells, which were respectively cultured in Dulbecco's Modified Eagle Medium (DMEM) and Roswell Park Memorial Institute 1640 (RPMI-1640) supplemented with 10% fetal bovine serum and incubated under 37 ℃ and 5% CO2.
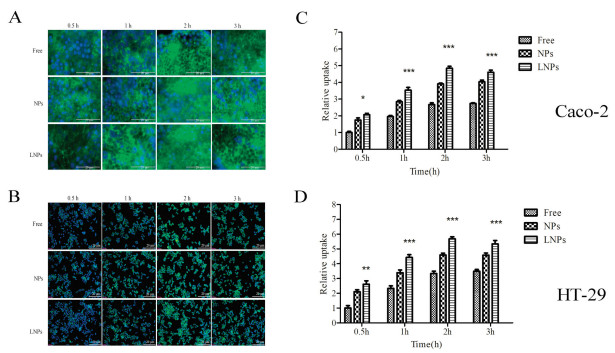
Coumarin 6 (Cou-6, green channel) was chosen as a fluorescent probe, instead of VP5, to detect the uptake of LNPs by Caco-2 cells and HT-29 cells using HCS (Thermo Scientific Cellomics, Thermo, USA) and flow cytometric (FCM). To evaluate the uptake profiles, Caco-2 and HT-29 cells at an initial density of 10 × 104 cells per well were seeded in 24-well plates. The original medium was replaced by fresh medium containing free Cou-6, Cou-6 loaded NPs or coreshell LNPs at a final Cou-6 concentration of 20 ng/mL for 0.5, 1, 2, 3 h at 37 ℃, respectively. At the given time point, the media was removed and then cells were washed twice with cold PBS. Cells were fixed with 4% paraformaldehyde for 12 min at room temperature followed by washing with PBS for three times. The cells nuclei were then stained with Hoechst 33258 (2.5 μg/mL, blue channel) for 8 min. The cells were finally washed three times with PBS and imaged by HCS as described previously [39]. For FCM analysis, cells were collected and washed by PBS. Then intracellular Cou-6 fluorescence was analyzed by FCM after excitation with a 488 nm argon laser. Fluorescence emission at 525 nm from 20, 000 cells were collected to generate a single parameter histogram.
As shown in Fig. 3, all of free Cou-6, NPs and LNPs performed a time dependence behavior of uptake with the plateau period at 2 h. The dim green fluorescence was observed in the cytosol of Caco-2 cells (Fig. 3A) and HT-29 cells (Fig. 3B) in free Cou-6 group. NPs could rapidly accumulate in the cytosol of cells, which was revealed by bright green fluorescence. Furthermore, LNPs could further improve the efficiency of cell uptake after 2 h of incubation, which might result from the specific biocompatibility provided by the lipid membrane [5]. The enhanced cellular uptake of LNPs was also confirmed by the quantitative profiles by FCM (Figs. 3C and D). The fluorescence intensity of cells treated with LNPs was much stronger than that in other treated groups. Though the Cou-6-NPs was not the same with VP5-NPs (the VP5 was packed in the inter water phase while the Cou-6 was packed in the PLGA oil phase), it is still can verify the truth that the lipid nanoparticles were superior than the bare PLGA nanoparticles in the cellar uptake. Over all, the data of cellar uptake demonstrated that more Cou-6- LNPs entered cells than Cou-6-NPs on both cell lines, indicating a better affinity of LNPs with intestinal epithelial cell membrane.

|
Download:
|
| Fig. 3. Uptake of LNPs by Caco-2 cells (A, C) and HT-29 cells (B, D). Scale bar: 20 μm. Data are expressed as mean ± SD (n = 3). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. LNPs/NPs. | |
The tight junction of the intestinal epithelium provides a physical barrier to limit the uptake of the macromolecules such as peptides [40]. Even though the little bit of the peptides were absorbed, they still suffered the short half-life in circulation [41]. More difficult for oral peptide drug delivery was the low permeability of peptide when through the intestinal epithelium membrane [42]. Changing the charge or particle size of nanoparticles, however, is only one of the two benefits of increasing intracellular differentiation or permeation of biofilm. So far, there is no balanced approach. Semi-elastic core-shell nanoparticles, modified nanoparticles with lipid membranes might improve the peptide delivery efficiency to the intestinal epithelium membrane, which could provide more opportunities to penetrate the intestinal epithelium cell membrane and then enter the circulation. Encouraged by the results in cells that our design could improve the delivery efficiency to the intestinal epithelium, we attempt to test whether the semi-elastic core-shell nanoparticles could cause enhanced oral absorption of VP5.
Eight-week-old male Sprague-Dawley (SD) rats with weight between 180 g to 220 g were purchased from the Laboratory Animal Center of Sichuan University (Sichuan University, Chengdu, Sichuan, China). The experiments were approved and supervised by the State Key Laboratory of Biotherapy Animal Care and Use Committee (Sichuan University, Chengdu, Sichuan, China).
Rats were divided randomly into three groups for oral administration of free VP5, VP5-NPs and VP5-LNPs at the same dose of 1.2 mg/kg (n = 6 for each studied group), respectively. Before the experiment, all rats were fasted for 12 h with free access to water. Each rat was administered an oral volume of 0.5 mL/100 g. Approximate 250 μL of blood samples were collected from the orbit into a heparinized centrifuge tube before and at 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48 and 72 h post-dosing. All blood samples were centrifuged at 3500 rpm (4 ℃) for 10 min to obtain the plasma samples. The plasma samples were collected and stored at -20 ℃ until analyzed by high-performance liquid chromatography (HPLC). And the profiles were subsequently analyzed by Drug and Statistics 2.0 (DAS 2.0) (Mathematical Pharmacology Professional Committee of China, Shanghai, China). The obtained data are reported as mean ± SD. Inter-group differences were statistically analyzed by an unpaired, two-tailed Student's t test (Graph Pad Prism version 5.1) between NPs and LNPs group. P < 0.05 was considered a statistically difference. P < 0.01 was considered a statistically significant difference.
The pharmacokinetic profiles were illustrated in Fig. 4 and the critical pharmacokinetic parameters were listed in Table 1. Amazingly, these results coincided with that of cell experiment. VP5-LNPs of proper size could improve the intestinal absorption and subsequently reach the systemic circulation and gradually release VP5, which might enhance the bioavailability of VP5. The AUC0-72h values in the SD rats of VP5-LNPs were 2.55 folds in relative to those in VP5-NPs group. The t1/2z has improved 2.02 folds in the VP5-LNPs than those in the VP5-NPs group. Currently, the underlying mechanism for how and why semi-elastic coreshell nanoparticles enhances peptide delivery efficacy is still under investigation.We still need to investigate the biodistribution of our nano-preparations in intestinal tract. This is not an easy job because, in biological environments, it needs high sensitivity and spatiotemporal resolution to precisely visualize the microscopic movement of nanoparticles, especially their rotation and deformation, requires further quantification.

|
Download:
|
| Fig. 4. Plasma concentration-time curves from SD rats treated with free VP5, VP5-NPs and VP5-LNPs after a single gavage administration (n = 6). | |
|
|
Table 1 Pharmacokinetic parameters after a single gavage administration in SD rats. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
In summary, in this study, we pioneered semi-elastic core-shell nanoparticles for oral delivery of peptide. We systematically optimized and obtained optimal semi-elastic core-shell nanoparticles loaded VP5 with excellent entrapment efficiency and better particle size. Then, we evaluated its oral delivery efficiency by pharmacokinetics in vivo and cellular uptake in vitro. In vivo pharmacokinetic experiment showed that VP5-LNPs could improve the plasma concentration of VP5, indicating enhanced oral bioavailability. In vitro cellular uptake experiment showed that the semi-elastic core-shell VP5-LNPs could greatly improve the efficiency of uptake by intestinal epithelial cells, giving more chance for delivering to the circulation. This might be explained by the presence of lipid layer coating on nanoparticles. Overall, these data supported the belief that VP5-LNPs would be an excellent oral peptide delivery system and worthy of further investigation as a promising candidate for oral disease treatment. And this study may provide some methodological clues and insights for the design of oral peptide delivery system. However, there are still many underlying mechanisms of semi-elastic core-shell nanoparticles loaded peptide that have not been studied in depth. Hence, superresolution microscopy and molecular dynamics of the semi-elastic core-shell nanoparticles is worthy of being further investigated. We anticipate that these research results will be seen in the future and encourage the design of semi-elastic core-shell nanoparticles for peptide delivery.
AcknowledgmentsThis work was financially supported by the Science and Technology Project of Shenzhen (No. JCYJ20170413155047512) and Scientific Research Foundation of the Science and Technology Department of Sichuan Province, China (No. 2018JY0143). We thank Yuan Huang and her students from the West China Pharmaceutical College for providing us with the Caco-2 cell.
| [1] |
J.H. Hamman, G.M. Enslin, A.F. Kotzé, Biod. Clinic Immuno. Biopharm. Gene Ther. 19 (2005) 165-177. |
| [2] |
R.I. Mahato, A.S. Narang, L. Thoma, et al., Crit. Rev. Ther. Drug Carrier Syst. 20 (2003) 153-214. DOI:10.1615/CritRevTherDrugCarrierSyst.v20.i23.30 |
| [3] |
Z. Niu, I. Conejos-SÃnchez, B.T. Griffin, et al., Adv. Drug Deliv. Rev. 106 (2016) 337-354. DOI:10.1016/j.addr.2016.04.001 |
| [4] |
Y. Zhang, H. Li, Q. Wang, et al., Adv Funct Mater. (2018) 1802675. |
| [5] |
M. Yu, L. Xu, F. Tian, et al., Nat. Commun. 9 (2018) 2607. DOI:10.1038/s41467-018-05061-3 |
| [6] |
L.M. Ensign, R. Cone, J. Hanes, Adv Drug Deliv Rev. 64 (2014) 557-570. |
| [7] |
C. Salvador-Morales, L. Zhang, R. Langer, et al., Biomater. 30 (2009) 2231-2240. DOI:10.1016/j.biomaterials.2009.01.005 |
| [8] |
C. Heneweer, S.E. Gendy, O. Penate-Medina, Ther Deliv. 3 (2012) 645-656. DOI:10.4155/tde.12.38 |
| [9] |
H. Seyednejad, A.H. Ghassemi, C.F. van Nostrum, et al., J. Controll Release 152 (2011) 168-176. DOI:10.1016/j.jconrel.2010.12.016 |
| [10] |
A.K. Srivastava, P. Bhatnagar, M. Singh, et al., Int.J.Nanomed. 8 (2013) 1451-1462. |
| [11] |
W. Shan, X. Zhu, M. Liu, et al., ACS Nano 9 (2015) 2345-2356. DOI:10.1021/acsnano.5b00028 |
| [12] |
W. Shan, X. Zhu, W. Tao, et al., ACS Appl. Mater. Interfaces 8 (2016) 25444-25453. DOI:10.1021/acsami.6b08183 |
| [13] |
T. Yu, S. Zhao, Z. Li, et al., Int. J. Mol. Sci. 17 (2016) 1977. DOI:10.3390/ijms17121977 |
| [14] |
C.P. Reis, R.J. Neufeld, A.J. Ribeiro, et al., Nanomed. 2 (2006) 8-21. DOI:10.1016/j.nano.2005.12.003 |
| [15] |
Q. Li, D. Xia, J. Tao, et al., J. Pharm. Sci. 106 (2017) 3120-3130. DOI:10.1016/j.xphs.2017.05.029 |
| [16] |
S.H. Kang, V. Revuri, S.J. Lee, et al., ACS Nano 11 (2007) 10417-10429. |
| [17] |
K. Sonaje, K.J. Lin, J.J. Wang, et al., Adv. Funct. Mater. 2005 (2010) 3695-3700. |
| [18] |
S. Zhao, J. Li, Y. Zhou, et al., Front. Pharmacol. 4 (2019) 74-83. |
| [19] |
Y. Liu, J. Pan, S.S. Feng, J. Pharm. Sci. 395 (2010) 243-250. |
| [20] |
P.J. Stevens, M. Sekido, R.J. Lee, Pharm Res. 21 (2004) 2153-2157. DOI:10.1007/s11095-004-7667-5 |
| [21] |
S.C. Semple, A. Chonn, P.R. Cullis, Biochem. 35 (1996) 2521-2525. DOI:10.1021/bi950414i |
| [22] |
H.Y. Sun, K.W. Fang, D. Liu, Chin. J. Hypertens. 18 (2010) 91-94. |
| [23] |
S. Acharya, S.K. Sahoo, Adv Drug Deliv Rev. 63 (2011) 170-183. DOI:10.1016/j.addr.2010.10.008 |
| [24] |
M. García-Díaz, C. Foged, H.M. Nielsen, Int. J. Pharm. 482 (2015) 84-91. DOI:10.1016/j.ijpharm.2014.11.047 |
| [25] |
Y. Yang, Y. Yin, J. Zhang, et al., Pharmaceutics 10 (2018) 146. DOI:10.3390/pharmaceutics10030146 |
| [26] |
A. Lalatsa, V. Lee, J.P. Malkinson, et al., Mol. Pharm. 9 (2012) 1665-1680. DOI:10.1021/mp300009u |
| [27] |
A.T. Florence, A.M. Hillery, N. Hussain, et al., J. ControlRelease 36 (1995) 39-46. |
| [28] |
T. Ma, L. Wang, Y. Ting, et al., Colloids Surf. B:Biointerfaces 117 (2014) 512-519. DOI:10.1016/j.colsurfb.2014.02.039 |
| [29] |
Y. Fei, L. Yang, S.L. Chang, et al., Int. J. Pharm. 484 (2015) 181-191. DOI:10.1016/j.ijpharm.2015.02.055 |
| [30] |
X. Song, Z. Yu, W. Wu, et al., Int. J. Pharm. 350 (2008) 320-329. DOI:10.1016/j.ijpharm.2007.08.034 |
| [31] |
F. Yu, M. Ao, X. Zheng, et al., Drug Deliv. 24 (2017) 825. DOI:10.1080/10717544.2017.1321062 |
| [32] |
A. Zafar, J. Ali, A. Bhatnagar, et al., Int. J. Biol. Macromol. 65 (2014) 479-491. DOI:10.1016/j.ijbiomac.2014.02.002 |
| [33] |
R.M. Hathout, S. Mansour, N.D. Mortada, et al., Aaps. Pharmscitech. 8 (2007) E1-E12. |
| [34] |
D.L. Fang, Y. Chen, B. Xu, et al., Int. J. Mol. Sci. 15 (2014) 3373-3388. DOI:10.3390/ijms15033373 |
| [35] |
C.T. Turk, U.C. Oz, T.M. Serim, C. Hascicek, AAPSPharm. Sci.Tech. 15 (2014) 161-176. DOI:10.1208/s12249-013-0048-9 |
| [36] |
J. Li, Z. He, S. Yu, et al., J. Biomed. Nanotechnol. 8 (2012) 809-817. DOI:10.1166/jbn.2012.1433 |
| [37] |
L. Mu, S. Feng, J. Control Release 86 (2003) 33-48. DOI:10.1016/S0168-3659(02)00320-6 |
| [38] |
Z. Dong, S. Xie, L. Zhu, et al., Drug Deliv. 18 (2011) 441. DOI:10.3109/10717544.2011.577109 |
| [39] |
Q.X. Song, M. Huang, L. Yao, et al., ACS Nano 8 (2014) 2345-2359. DOI:10.1021/nn4058215 |
| [40] |
L. Plapied, N. Duhem, A.D. Rieux, et al., Curr. Opin. Colloid. Interf. Sci. 16 (2011) 228-237. DOI:10.1016/j.cocis.2010.12.005 |
| [41] |
S. Frokjaer, D.E. Otzen, Nat. Rev. Drug Discov. 4 (2005) 298. DOI:10.1038/nrd1695 |
| [42] |
K. Maisel, L. Ensign, M. Reddy, et al., J. Controll Release 197 (2015) 48-57. DOI:10.1016/j.jconrel.2014.10.026 |