 2020, Vol. 31
2020, Vol. 31 


b Advanced Institutes of Convergence Technology, Gyeonggi-do 16229, Republic of Korea
Protein, one of the most important biopolymers in nature, plays many important roles in life for its unique structure-specific function in a certain environment. In a living system, proteins form supra-molecular structures through secondary bonds to transport biomaterials across cellular membranes, construct networks of cytoskeleton, and regulate enzymatic reactions [1-3]. Supramolecular assembly of proteins thus has been extensively studied by many researchers worldwide to construct highly ordered protein nanostructures, using peptide tags, metal ion interactions, and other chemical bonds [4-8], for various biomedical applications such as biosensing [9, 10], and drug delivery [11, 12]. However, such secondary binding strategies involve sophisticated interactions and often cause a high degree of disorder in the final assembly of protein nanostructures. Hence, a number of research groups have introduced DNAs as binding motifs to facilitate one, two, and three-dimensional protein nanostructures by precisely controlling supra-molecular bonds through Watson-Crick baseparing of DNAs [13-19].
We combine avidin-biotin conjugate system and sequence specific base-pairing of DNA to form polymerized arrays of traptavidins (TAv's). TAv has exactly four biotin binding sites and easily bind to biotinylated moieties through avidin-biotin coupling, one of the strongest secondary bonds in nature [20]. Furthermore, TAv has excellent thermal and mechanical stabilities, and stronger binding affinity to biotinylated molecules than that of streptavidin (SAv). Here we develop a simple method to construct pre-programmed linear nanostructures by biochemically combining TAv's and DNAs through step-growth polymerization approach [21]. Using this molecular assembly method, we have constructed extended nanostructures with linear chains of hybrid biopolymers and also shown linear arrays of gold nanoparticles through sitespecific avidin-biotin interactions [22, 23].
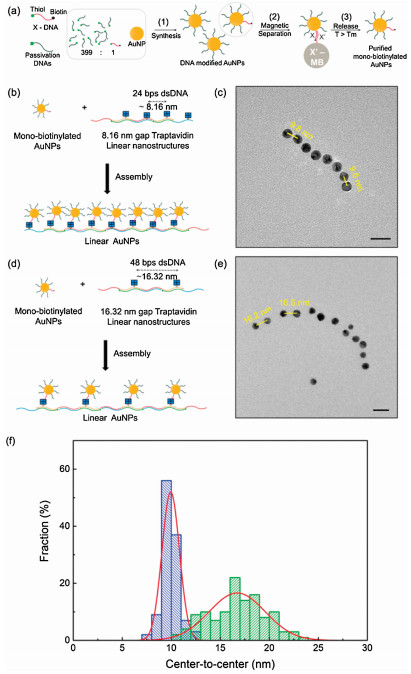
Fig. 1a is the scheme for TAv-DNA monomer synthesis. We designed 48 base pair (bp) DNA strands modified with double biotin at internal thymine of the 20th base location to tether TAv with two nearby biotin binding sites out of 4 binding sites of TAv. To synthesize TAv-DNA monomers, we reacted excess TAv and double-biotinylated DNAs in PBS buffer at a molar ratio of 10:1. In order to purify only TAv-DNA conjugates from mixtures, a magnetic separation method was conducted. We used commercially available magnetic beads (MBs) with in-house surface functionalization for the purification process. The capture DNAs are complementary to the target DNAs and bound to the surface of the MBs via highly stable covalent bonds based on maleimide - thiol chemistry [24]. Then, the mixture was attached to the surface of 15 bp A complementary-DNA modified magnetic beads (A'-MBs) that enable us to capture TAv-DNA monomers in a sequence-specific manner, and TAv without the doublebiotinylated DNA strands were washed out. The melting temperature (Tm) of DNA is determined by G-C contents, DNA concentration, and salt environment [25]. In our case, samples are captured by 15 bps DNA probes which of melting point is about 55 ℃ in PBS buffer. TAv-DNA monomers were dehybridized from the surface of A'-MB by thermal release at 60 ℃ for 5 min. After magnetic separation, about 91% of the separation yield was obtained in the measurement of solution absorbance at 260 nm of DNA (Fig. S1 in Supporting information). This is because TAv is strongly bound to biotin at the releasing temperature. TAv-DNA monomers could be easily obtained because the binding affinity between TAv and biotinylated DNA was stable up to 70 ℃ [20, 26].

|
Download:
|
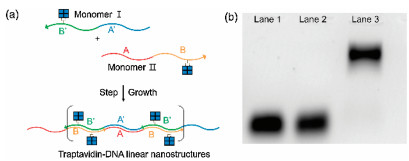
| Fig. 1. (a) Schemes for synthesizing traptavidin-DNA monomers with magnetic separation: (1) mixing TAv with bis-biotinylated DNA at molar ration of 10:1, (2) separating the TAv tethered A-DNA using A0-MBs, (3) removing unreacted TAvs, (4) releasing TAv-DNA complexes from A0-MBs. (b) 12% PAGE analysis for TAv-DNA complexes. Lane 1 to lane 6: 20:1, 10:1, 5:1, 2.5:1, 1:1 and 1:2 (molar ratio of TAv and DNA). Lane 7 and Lane 8 are TAv-DNA conjugates after magnetic separation after mixing TAv with DNA at molar ratio of 20:1. | |
{kind=link}
Fig. 1b shows the results of 12% polyacrylamide gel electrophoresis (PAGE) analysis to determine the relative molecular weights of TAv-DNA conjugates according to the molar ratio of TAv and double-biotinylated DNA. Lane 1 to lane 6 are the results of reactions in which the amounts of TAv's are successively decreased relative to the amount of double-biotinylated DNAs. The results show that the amount of conjugates with two doublebiotinylated DNAs increases as the amount of TAv decreases. In lane 5, where the molar ratio of TAv and double-biotinylated DNA is 1:1, the dominant band shows lower mobility because the conjugates with two DNAs have higher molecular weight than the conjugates with single DNAs. Based on these experimental conditions, TAv-single DNA are synthesized by reacting Traptavidin and DNA at a molar ratio of 10:1 in order to obtain a conjugate having only one double-biotinylated DNA in TAv multivalent core. Lane 7 and lane 8 are monomer A and monomer B, respectively, purified by magnetic separation method as shown in Fig. 1a. From the result of band positions in electrophoresis, we confirm that only the conjugates with one double-biotinylated DNA are selectively obtained.
To proceed with DNA-mediated TAv polymerization, we designed two distinct monomer sets that can be assembled to polymers through the sequence complementarity at both ends of each DNA strands. The 48 bp monomer Ⅰ consists of 24 bp A-DNA sequence complementary to the 24 bp A0-DNA sequence of 48 bp monomer Ⅱ. Also, the monomer Ⅰ consists of 24 bp B-DNA sequence which is complementary to the 24 bp B0-DNA sequence of monomer Ⅱ. Fig. 2a is a scheme for DNA-directed step-growth method using the monomers Ⅰ and Ⅱ. The monomers were mixed at the molar ratio of 1:1 in 0.5 mol/L NaCl PBS buffer. The mixture was reacted at 60 ℃ for 10 min and cooled down to RT for 12 h. Fig. 2b shows the results of 1% agarose electrophoresis. Lane 1 and Lane 2 are units of monomers Ⅰ and Ⅱ, respectively. Lane 3 is TAv-DNA linear nanostructure polymerized by the step-growth method. Since the molecular weight is larger than that of monomers, TAvDNA linear nanostructure showed that the position of the band is above the monomers.

|
Download:
|
| Fig. 2. (a) Scheme for polymerizing TAv-DNA linear nanostructure via pre-designed DNA hybridization. (b) Result of 2% agarose gel electrophoresis. Lanes 1 and 2 are Traptavidin-DNA monomers Ⅰ and Ⅱ. Lane 3 is TAv-DNA linear chain after hybridizing monomers Ⅰ and Ⅱ. | |
{kind=link}
We synthesized clustered nanostructures based on TAv-DNA linear nanostructure and biotinylated DNA modified gold nanoparticles (AuNPs). DNA functionalization to the surface of 8 nm AuNPs was conducted by salt-aging method [27]. We designed the DNA strands with thiol modification at the 5' end of DNA to functionalize the surface of AuNPs, and biotin modification at the 3' end of DNA to couple to the biotin binding site of TAv on the linear nanostructure. As a result of UV–vis absorption spectroscopy, the maximum peak of citrate-stabilized 8 nm AuNPs was 518.65 nm and 523.52 nm for DNA modified AuNPs [28] (Fig. S2 in Supporting information). The length of 48 bp dsDNA template is about 16.32 nm and the spacing between the two TAv's is about 8.16 nm. Then, the biotinylated AuNPs were assembled to form clustered AuNPs by conjugating biotinylated DNAs on the surface of AuNPs and biotin binding sites of TAv on the linear nanostructures, as shown in Fig. 3a. Fig. 3b is a TEM image of AuNPs without the TAv-DNA linear nanostructures. Fig. 3c is the TEM image of clustered AuNPs assembled with biotinylated DNAs on AuNPs and TAv's on the linear nanostructures. Also, SEM images were also obtained (Fig. S5a in Supporting information).

|
Download:
|
| Fig. 3. (a) Schemes for clustered plasmonic Au nanostructures. Biotinylated DNA modified 8 nm AuNPs were assembled with TAv-DNA linear nanostructures. (b) TEM images of biotinylated DNA modified 8 nm AuNPs unreacted with TAv linear nanostructures a non-optimized assembly reaction. (c) TEM images of clustered AuNPs after mixing biotinylated DNA modified AuNPs with TAv linear nanostructures, scale bar =100 nm. | |
{kind=link}
Using the TAv-DNA linear nanostructures as templates, we construct one-dimensional arrays of plasmonic AuNPs with passivation DNAs to prevent gold clustering. The maximum number of thiolated DNAs bound to the surface of 15 nm AuNPs is about 240 under a salt-aging condition of about 0.5 mol/L NaCl [29]. In order to prevent clustering of nanostructures, we limited the number of biotinylated X-DNA on the surface of 8 nm AuNPs by surface passivation with non-biotinylated DNAs [30]. Although there is no guarantee that only one biotinylated DNA is attached to the surface of 8 nm AuNPs, it is highly probable that "monobiotinylated" AuNPs dominates the population, because the relatively small surface areas of 8 nm AuNPs are passivated with a mixture of non-biotinylated DNAs and biotinylated DNAs at a high molar ratio of 399:1.
The magnetic separation method using X'-MBs was applied to remove AuNPs that are covered with passivation DNA only. After unpurified AuNPs and X'-MBs were mixed in a PBS buffer solution for 3 h, the AuNPs without biotinylated X-DNAs were washed out from X'-MBs. Subsequently, only AuNPs with biotinylated X-DNA were selectively collected and thermally released at 60 ℃ for 5 min, as shown in Fig. 4a. To determine the number of DNAs bound to AuNP, DNAs bound to AuNPs was reduced by Dithiothreitol (DTT) solution and quantified by the fluorescence intensities of DNA bound to AuNPs. We used Cy3 labeled DNAs instead of biotin to determine how many numbers of biotins attached to the surface of AuNP depending on the ratio of passivation DNAs and biotnylated DNAs. An average number of Cy3 labeled DNA was ~61.80, and the number of cy3 of AuNPs mixed with 399:1 ratio of passivation DNA and cy3-labeled DNA was about 1.39 per AuNP (Fig. S4 in Supporting information). After the mono-biotinylated AuNP was purified, TAv's on the linear nanstructures and mono-biotinylated AuNPs were assembled at a molar ratio of 1:1, as shown in Fig. 4b. Fig. 4c is the TEM image of linear plasmonic AuNP nanostructues with inter-particle spacing of about 9.9 nm. In the same manner, we synthesized TAv linear nanostructures with TAv-conjugated monomer Ⅰ and TAv-unconjugated monomer Ⅱ, as shown in Fig. 4d. Also, SEM images were also obtained (Fig. S5b in Supporting information). TEM images in Fig. 4e showed that the AuNPs were separated at about 16.7nm that is consistent with the design scheme in Fig. 4d. As a result, it was confirmed that the AuNPs were well conjugated along the linear DNA chain through uranyl acetate stained TEM image (Fig. S5). The distribution of center-to-center distance of AuNP arrays was displayed with representative histograms in Fig. 4f. Gaussian fit was used to quantify the standard deviation of center-to-center distance, resulting that the standard deviation of TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-conjugated monomer Ⅱ is 0.89 and of TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-unconjugated monomer Ⅱ is 2.85. The statistical analysis matches well with the design of the nano-templates based on TAvDNA conjugates with predetermined inter-TAv separations.

|
Download:
|
| Fig. 4. (a) Schemes for synthesizing mono-biotinylated DNAs onto AuNPs: (1) mixing passivation DNA and biotinylated DNA (molar ration of 399:1) with AuNPs using salting aging method; (2) separating mono-biotinylated X-DNA modified AuNPs through X'-MB; (3) releasing the AuNPs from the surface of X'-MBs. (b) Mono-biotinylated AuNPs were assembled with conjugated with TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-conjugated monomer Ⅱ. (c) TEM imagesof AuNP arraysseparated at ~9.9nm gap distance, scale bar = 20nm. (d) Mono-biotinylated AuNPs were conjugated with TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-unconjugated monomer Ⅱ. (e) TEM images of AuNP arrays separated at ~16.7nm gap distance, scale bar = 20nm. (f) Analysis of the center-to-center distances of AuNP arrays from TEM images. Blue histogram for center-to-center distances of AuNP arrays conjugated to TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-conjugated monomer Ⅱ. Green histogram of center-to-center distances of AuNP arrays conjugated to TAv DNA linear nanostructures assembled with TAv-conjugated monomer Ⅰ and TAv-unconjugated monomer Ⅱ. The line curve is a Gaussian fit to the histogram. | |
{kind=link}
In conclusion, we have introduced step-growth polymerization to construct a linear array of supramolecular building blocks made of traptavidins and DNAs through simple magnetic separation technique. Two different sets of traptavidin-DNA conjugates are connected in turn to make one-dimensional assembly of the protein-DNA complex polymers. Spacings of the constituent proteins are predetermined by designing the base sequence of the DNAs in the building blocks. The linear array of the proteinDNA complex polymers are used as one-dimensional templates to precisely locate gold nanoparticles at the TAv sites with excess valences, resulting in plasmonic nanostructures with predetermined inter-particle spacings. Such plasmonic nanostructures are expected to increase optical signals for fluorescence-based biosensing and bioimaging applications [31-34]. Furthermore, the linear nano-templates of TAv-DNA conjugates could be readily extended to various nanomaterials, both organic and inorganic ones, through precisely controlled, periodically expressed binding sites, for a wide range of biomedical applications from molecular diagnostics to smart drug delivery [35-38].
AcknowledgmentThis work was supported by the Brain Research Program (No. 2016M3C7A1904987) through the National Research Foundation of Korea (NRF).
Appendix A. Supplementary dataSupplementary material related to this article can be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.07.008.
| [1] |
L. Artzi, E.A. Bayer, S. Morais, Nat. Rev. Microbiol. 15 (2017) 83-95. DOI:10.1038/nrmicro.2016.164 |
| [2] |
T.D. Pollard, G.G. Borisy, Cell 112 (2003) 453-465. DOI:10.1016/S0092-8674(03)00120-X |
| [3] |
S. Wang, H. Arellano-Santoyo, P.A. Combs, J.W. Shaevitz, Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 9182-9185. DOI:10.1073/pnas.0911517107 |
| [4] |
B. An, X. Wang, M. Cui, et al., ACS Nano 11 (2017) 6985-6995. DOI:10.1021/acsnano.7b02298 |
| [5] |
J.D. Brodin, X.I. Ambroggio, C.Y. Tang, et al., Nat. Chem. 4 (2012) 375-382. DOI:10.1038/nchem.1290 |
| [6] |
J.C.T. Carlson, S.S. Jena, M. Flenniken, et al., J.Am.Chem.Soc. 128 (2006) 7630-7638. DOI:10.1021/ja060631e |
| [7] |
Q. Qi, T.X. Zhao, B.L. An, X.Y. Liu, C. Zhong, Chin.Chem.Lett. 28 (2017) 1062-1068. DOI:10.1016/j.cclet.2016.12.008 |
| [8] |
F. Sun, W.B. Zhang, Chin. Chem. Lett. 28 (2017) 2078-2084. DOI:10.1016/j.cclet.2017.08.052 |
| [9] |
C.H. Chen, X.H. Ji, Chin. Chem. Lett. 29 (2018) 1287-1290. DOI:10.1016/j.cclet.2017.10.013 |
| [10] |
M. Raeeszadeh-Sarmazdeh, E. Hartzell, J.V. Price, W. Chen, Curr. Opin. Chem. Eng. 13 (2016) 109-118. DOI:10.1016/j.coche.2016.08.016 |
| [11] |
Y. Hsia, J.B. Bale, S. Gonen, et al., Nature 535 (2016) 136-139. DOI:10.1038/nature18010 |
| [12] |
E.J. Lee, N.K. Lee, I.S. Kim, Adv. Drug Deliv. Rev. 106 (2016) 157-171. DOI:10.1016/j.addr.2016.03.002 |
| [13] |
P.K. Lo, K.L. Metera, H.F. Sleiman, Curr. Opin. Chem. Biol. 14 (2010) 597-607. DOI:10.1016/j.cbpa.2010.08.002 |
| [14] |
D. Wang, J. Song, P. Wang, et al., Nat. Protoc. 13 (2018) 2312-2329. DOI:10.1038/s41596-018-0039-0 |
| [15] |
F. Wang, B. Willner, I. Willner, Curr. Opin. Biotechnol. 24 (2013) 562-574. DOI:10.1016/j.copbio.2013.02.005 |
| [16] |
P. Wang, S. Gaitanaros, S. Lee, et al., J. Am. Chem. Soc. 138 (2016) 7733-7740. DOI:10.1021/jacs.6b03966 |
| [17] |
X.R. Wu, C.W. Wu, F. Ding, et al., Chin. Chem. Lett. 28 (2017) 851-856. DOI:10.1016/j.cclet.2017.01.012 |
| [18] |
H. Zhang, Y. Wang, H. Zhang, et al., Nat. Commun. 10 (2019) 1006. DOI:10.1038/s41467-019-09004-4 |
| [19] |
T. Zhang, C. Hartl, K. Frank, et al., Adv. Mater. 30 (2018) e1800273. DOI:10.1002/adma.201800273 |
| [20] |
C.E. Chivers, E. Crozat, C. Chu, et al., Nat. Methods 7 (2010) 391-393. DOI:10.1038/nmeth.1450 |
| [21] |
J.R. McMillan, O.G. Hayes, J.P. Remis, C.A. Mirkin, J. Am. Chem. Soc. 140 (2018) 15950-15956. DOI:10.1021/jacs.8b10011 |
| [22] |
W.X. Ren, J.Y. Han, S. Uhm, et al., Chem.Commun.(Camb.) 51 (2015) 10403-10418. DOI:10.1039/C5CC03075G |
| [23] |
J.Y. Song, W.T. He, H. Shen, et al., Chem. Commun. (Camb.) 55 (2019) 2449-2452. DOI:10.1039/C8CC09717H |
| [24] |
X. Xu, N.L. Rosi, Y. Wang, F. Huo, C.A. Mirkin, J. Am. Chem. Soc. 128 (2006) 9286-9287. DOI:10.1021/ja061980b |
| [25] |
S.M. Freier, R. Kierzek, J.A. Jaeger, et al., Proc. Natl. Acad. Sci. U. S. A. 83 (1986) 9373-9377. DOI:10.1073/pnas.83.24.9373 |
| [26] |
Y.Y. Kim, Y. Bang, A.H. Lee, Y.K. Song, ACS Nano 13 (2019) 1183-1194. DOI:10.1021/acsnano.8b06170 |
| [27] |
H.D. Hill, C.A. Mirkin, Nat. Protoc. 1 (2006) 324-336. DOI:10.1038/nprot.2006.51 |
| [28] |
K.G. Witten, J.C. Bretschneider, T. Eckert, W. Richtering, U. Simon, Phys. Chem. Chem. Phys. 10 (2008) 1870-1875. DOI:10.1039/b719762d |
| [29] |
S.J. Hurst, A.K. Lytton-Jean, C.A. Mirkin, Anal. Chem. 78 (2006) 8313-8318. DOI:10.1021/ac0613582 |
| [30] |
D.K. Lim, K.S. Jeon, H.M. Kim, J.M. Nam, Y.D. Suh, Nat. Mater. 9 (2010) 60-67. DOI:10.1038/nmat2596 |
| [31] |
Y. Huang, P. Huang, J. Lin, Small Methods 3 (2019) 1800394. DOI:10.1002/smtd.201800394 |
| [32] |
A. Klinkova, R.M. Choueiri, E. Kumacheva, Chem. Soc. Rev. 43 (2014) 3976-3991. DOI:10.1039/c3cs60341e |
| [33] |
K. Vogele, J. List, G. Pardatscher, et al., ACS Nano 10 (2016) 11377-11384. DOI:10.1021/acsnano.6b06635 |
| [34] |
J. Mosquera, Y. Zhao, H.J. Jang, et al., Adv. Funct. Mater. (2019) 1902082. |
| [35] |
L.Y. Chou, K. Zagorovsky, W.C. Chan, Nat. Nanotechnol. 9 (2014) 148-155. DOI:10.1038/nnano.2013.309 |
| [36] |
P.A. Rasheed, N. Sandhyarani, Analyst 140 (2015) 2713-2718. DOI:10.1039/C5AN00004A |
| [37] |
H. Xue, F. Ding, J. Zhang, et al., Chem. Commun. (Camb.) 55 (2019) 4222-4225. DOI:10.1039/C9CC00175A |
| [38] |
L. Zhang, S.R. Jean, X. Li, et al., Nano Lett. 18 (2018) 6222-6228. DOI:10.1021/acs.nanolett.8b02263 |