 2020, Vol. 31
2020, Vol. 31 


The continuous development of optical-control cleavage reactions in the past few decades has offered a tremendous opportunity for the research and manipulation of metabolic processes in living systems [1-3]. Their capacity to provide spatial and temporal control to biological processes helps them attract lots of biochemists. Photocaging protein can be expressed using genetically encoded caged amino acids [4, 5], such as caged Cas9, When K866 was photocaged it was completely inactive before UV illumination [4]. By means of nucleic acid solid-phase synthesis, photo-cleavage molecule was incorporated into oligonucleotides that hybridized with an aptamer, which resulted in inactivation of the aptamer in the absence of light triggering [6]. Photocaging group was directly modified at the nucleobase of aptamers, DNAzymes, antisense agents and DNA decoy to regulate their activity using light [7-9]. When C1OH of unnatural azido sugars was photocaged, its metabolic labeling process was blocked, leading no azido group introduced onto the cell surface [10]. Besides the hydroxyl group, there is photolabile protecting group for the formyl group of 5fC preventing the cross-links of DNA and Histone [11]. Photocaging groups can be installed on many small molecules with biological activity and simple structure, such as the drug that alters the catalytic activity of DNA methyltransferases [12].
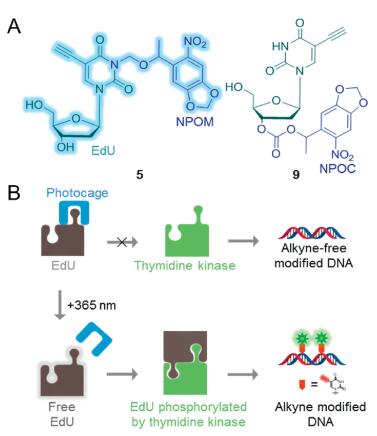
A series of bioorthogonal cleavage reactions has aroused, which have extended the bioorthogonal chemistry repertoire, capacitating an array of thrilling new biological applications [13]. We designed two photocaged 5-ethynyl-2'-deoxyuridine (EdU) derivatives to systematically investigate the effect of bond-cleavage type and position on the metabolic pathway of the EdU derivatives. We attached the N-3 position of the EdU base to 6-nitropiperonyloxymethyl (NPOM) and oxygen atom of deoxyribose 3' position to 6-nitropiperonyloxycarbonate (NPOC), which would be cleaved by irradiationwith 365 nm ultraviolet light, due to the optical activityof NPOM and NPOC groups (Fig. 1A).

|
Download:
|
| Fig. 1. Photocaging derivatives of EdU designed to optically modulate the metabolism of EdU in cells. (A) Caged EdU derivatives 5 and 9. (B) Scheme illustrating the principle of the photocaging approach. Photocaging derivatives of EdU cannot involve in nucleotide synthesis via salvage pathway, but after photoactivation by 365 nm, genomic DNA will be modified with alkyne by EdU released. | |
{kind=link}
EdU, a 2'-deoxythymidine (dT) analogue, is readily transported and incorporated into cellular DNA by cell metabolism. In addition, the terminal alkyne group at its C-5 position enable rapid and bioorthogonal cycloaddition of Cu(I)-catalyzed azide-alkyne socalled 'click reaction' [14]. NPOM installed on N-3 position of dT disrupt Watson-Crick base pairing [15-17]. When the caged dT was incorporated into the DNAzyme by DNA solid-phase synthesis, the more caged dT the chain contained, the lower the activity of DNAzyme [16], which indicated caged dT have the potential to be rejected by genomic DNA. Other than Watson-Crick base pairing, enzymatic reactions in vivo are highly specific. Previous reports have shown that dT with modifications atboth the 3' and C-5positionhad lower activity for thymidine kinase than their corresponding compound with only one modification [18]. Therefore, besides the N-3 photocaged EdU, we attached the photocaged group to the 3' position. After 3-(6-nitropiperonyloxymethyl)-5-ethynyl-2'-deoxyuridine (5) and 3'-(6-nitropiperonyloxycarbonate)-5-ethynyl-20-deoxyuridine (9) photodecageing with irradiation at 365 nm, the EdU is released and incorporated into cellular DNA during DNA replication. Whereafter, the genomic DNA is modified by alkyne groups, and the genome with alkyne groups is fluorescently labelled by click reaction (Fig. 1B).
After the identification of EdU derivatives 5 and 9, we evaluated their decaging capacity in vitro. Methanol is a benign solvent for 5 and 9. Besides, both of methanol and water are polar protic solvents, which can partly simulate physiological conditions. Most importantly, unlike DMSO, methanol solvents can be directly analyzed by HPLC (high performance liquid chromatography), and 3-nitro-2-ethyldibenzofuran caging group decaging capacity is also tested in methanol [19]. The result indicated that decaging capacity, by contrast, was influenced by which site of EdU the chromophore was attached. The amount of EdU released at 10 min is similar (Fig. 2), however, half-life of 5 is obviously shorter than that of 9 (Fig. 3A). Especially 5, photolysis could release more than 50% of EdU in 1 min, while 9 can only release about 25%, which was because the maximum absorption wavelength of 5 is closer to 365 nm (Fig. 3B). It is noteworthy that when 5 photolysis lasts for 2 min, the release of EdU does not change much (Fig. 3A). This may be because it produces an intermediate (Fig. 2A), which interfered photolysis efficiency of 5. We speculated that it was another photocaged EdU from the beginning. However, EdU did not increase along with the reduction of the intermediate after irradiating 5 at 365 nm for 1 min followed by keeping the system at 20 ℃ for one day. Moreover, heat from UV-lamp, rather than ultraviolet, allowed the intermediate to react (Fig. S1A in Supporting information). The intermediate may well be 1-(6-nitrosobenzo[d][1, 3]dioxol-5-yl)ethan-1-one after component separation and structure analysis for the 5+UV by HPLC and HRMS (Fig. S1B in Supporting information).

|
Download:
|
| Fig. 2. HPLC traced for the photocaging of 5 and 9 (15 μL, 2 mmol/L) at 0, 1, 2, 5, 8 and 10 minwith an elution system of 5%–100% acetonitrile inwater over 20 min at a flow rate of 1 mL/min, and the temperature was 35 ℃. (A) The initial peak retention time was 6.06 min. By comparison with the EdU standard, it was confirmed that compound 5 gradually dissociated and released EdU as the irradiation time increased.When the photodecaging for 1 min, the final peak in the signal is an intermediate producedduring the dissociation of compound 5. The intermediates will be photolyzed as the light time increases. (B) During the photodecaging of 9, no stable intermediate was produced except for EdU, but another major substance was produced, which was also produced during the dissociation of compound 5, indicating that it should be the leaving NPOM and NPOC. | |
{kind=link}

|
Download:
|
| Fig. 3. Photodecaging and spectroscopic analysis of 5 and 9. (A) is photodecaging curves and EdU release curves for compounds 5 and 9 when irradiated for 1, 2, 5, 8 and 10 min with 365 nm ultraviolet light. The left vertical axis is the normalized value of the integrated area of 5 and 9 detected by HPLC. The vertical axis on the right is the normalized value of the integrated area of the EdU released by 5 and 9 detected by HPLC. (B) UV–vis absorption spectra of EdU, NPOH (2), 5 and 9 in methanol, whose absorbance values were normalized. | |
{kind=link}
All of the experimental details for the synthesis, purifications and characterizations of the new compounds are listed in Supporting information. According to previous reports, the N-3 modification of dT requires protection of the two hydroxyl groups of deoxyribose and finally deprotection [16]. However, since NH is in α position of the two carbonyl groups, we speculate that NH should have a stronger acidity than the hydroxyl group. When an equivalent amount of a small base NaH is used, it can rapidly react with N—H to produce hydrogen and a nitrogen anion capable of resonating with the carbonyl group. It can be seen from the 2D NMR spectrum of compound 5 (in DMSO-d6) that NH disappeared in the low field and all of the hydroxyl groups could be found (Fig. S2 in Supporting information). When the photocage group was attached in the 3' position, the hydrogen of N-3 could be seen clearly in the low field (1H NMR of 9 in Supporting information). Surprisingly, the yield of conjugation of NPOM-Cl with deoxynucleoside is higher than that reported previously [16, 19], which may be the result of dynamics.
Photocaging on the EdU is not only due to its ability of incorporation into the genome, but also the low cytotoxicity of the molecule itself, which makes it less restrictive in intracellular applications. It was found by MTT experiments that 5 had little growth inhibition relative to the DMSO-treated control at 100 μmol/L for 24 h (Fig. S3 in Supporting information), which was 2 folds higher than that used in cell experiments. Therefore, the effects of compounds 5 and 9 on cell growth during cell experiments were negligible.
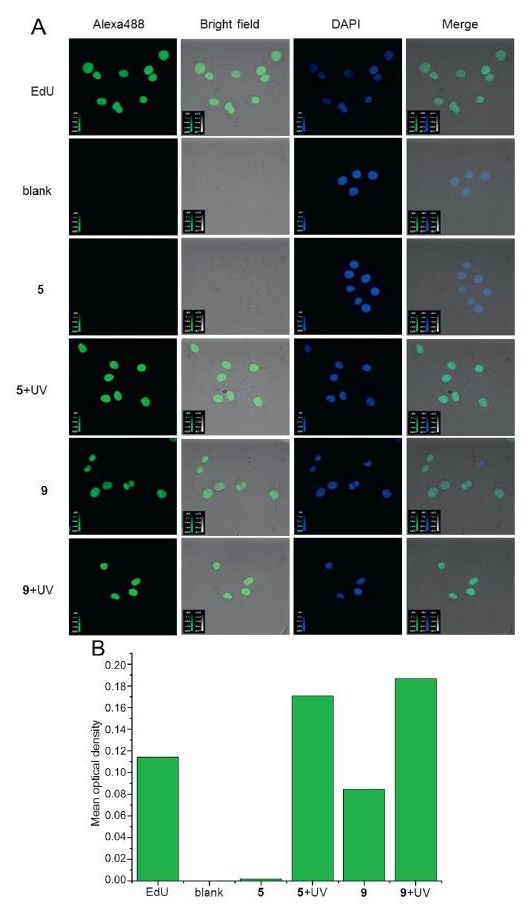
We incubated photocage-EdU 5 or 9 with cells and used cells treated only by PBS as blank experiment. Then the cells were fixed, permeabilized and stained with Alexa488 containing an azido group as well as DAPI followed by imaging with fluorescent confocal microscopy. As shown in Fig. 4, cells treated with compound 5 could not be stained by Alexa488, and the experimental results were similar to those of the blank experiment. However, cells treated with compound 9 could be stained by Alexa488 to green and overlapped with the blue stained by DAPI. The complete colocalization of the two colors indicating that 9 modified the genome with alkyne in the nucleus during incubation [20, 21]. Based on this result, we tested the stability of compound 9 in water. Compound 9 was dissolved in DMSO/water = 5/95 mixed solvent and incubated in the dark at 37 ℃ using an Incubator Shaker Series (Innova 42) for one day. The release of about 0.22% EdU was detected by HPLC, but no EdU was observed during the hydrolysis of compound 5 (Fig. S4 in Supporting information), which was consistent with the cell staining experiment. Although only 0.22% of the compound 9 decomposed, the concentration of EdU released was sufficient to incorporate into the genome and be stained, which can be supported by the previously reported literature [14]. To further explore the stability of 5 and 9, we conducted component separation by HPLC after shaking 5 and 9 in RIPA Lysis Buffer at 37 ℃ for one day. It produces the same result: Compound 5 has much better stability than 9. Unfortunately, both 5 and 9 produced EdU by their degradation in the buffer. In order to better contrast the dissociation rate of 5 and 9, we applied the linear fitting to calculating the equation of photometry working curve (Fig. S5 in Supporting information). Calculation results show that 9 exhibits a threefold dissociation rate than 5 (Table S1 in Supporting information).
We incubated EdU, 5 and 9 with cells separately, after cell adhesion, the cells treated with 5 and 9 were exposed to 365 nm ultraviolet radiation for 1 min and then cultured for 12 h. After that, the cells were fixed, permeabilized and stained with Alexa488 containing an azido group as well as DAPI followed by imaging with fluorescent confocal microscopy. As shown in Fig. 4A, after ultraviolet irradiation, cells, treated with compound 5 but not irradiation so that they could not be stained, were indeed stained and revealed complete colocalization with the secondary staining by DAPI, indicating that 5 could be photolysed normally under physiological conditions in cells and the results were similar to the cells treated with EdU (Fig. 4A). In Fig. 4B, mean optical density of cells treated by 5 and ultraviolet radiation were 100 folds higher than the cells only treated with 5 but not ultraviolet radiation. It is worth mentioning that, as for 5, ImageJ failed to track cell borders under optimal operation (Fig. 4B).

|
Download:
|
| Fig. 4. Fluorescence labeling of locus modified with alkyne groups. (A) HeLa cells were stained by Alexa488 and DAPI after chemical treatment, fixation and permeabilization. In the blank experiment, cells were treated with PBS only, and their nuclei were stained blue by DAPI only. The nuclei of cells treated by EdU can be stained green by Alexa488 and blue by DAPI. The cells treated with 5 had similar results as those in the blank experiment, but when the cells treated with 5 and were irradiated by 365 nm ultraviolet light for 1 min, the results were similar to those treated with EdU. Cells treated with 9 can be stained by two dyes regardless of whether they are irradiated or not. Merge means overlay of Alexa488 and DAPI channels, and revealed complete colocalization of the two colors. (B) Mean optical density of cells stained by Alexa488. | |
{kind=link}
Owing to the 3' and 5' positions of 5 having free hydroxyl groups that can be phosphorylated, and about half of 5 remaining after photolysis of 5 for 1 min, it is necessary to prove that the nuclei dyed into green were caused by the incorporation of EdU rather than 5 into genomic DNA. Therefore, genomic DNA was extracted from cells treated with compound 5 before and after ultraviolet irradiation of 365 nm. Genomic DNA of cells treated with PBS or EdU was blank experiment and a control experiment, respectively. Then we digested the genomic DNA into small molecules with enzymes and analyzed them by HPLC-MS, in addition, analyzed the DNA by dot blot experiments performed in vitro.
Liquid chromatography coupled to electrospray ionization tandem mass spectrometry analysis is often used to detect modified bases in the genomic DNA [22], and the signal can be amplified by chemical modification, but owing to EdU is artificially added, its amount in the genomic DNA is sufficient to be directly detected by mass spectrometry, although it carries an alkyne group which is prone to bioorthogonal reactions. The results of mass spectrometry analysis were similar to those of fluorescence confocal microscopy. Compound 5 and EdU were not detected in genomic DNA from the cells treated with compound 5, but only EdU was detected in genomic DNA from the cells treated with compound 5 and ultraviolet light (Fig. S6 in Supporting information), which indicating that alkyne of genome DNA was caused by EdU, not by compound 5.
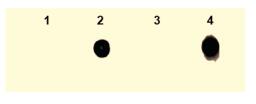
The interaction between biotin and avidin is a well-known noncovalent interaction, and it has a high affinity constant, so it has good sensitivity [23, 24]. After Click reaction between the extracted genomic DNA and Biotin-PEG4-N3 containing an azide modification, the DNA was adsorbed onto positively charged nylon membrane and dot blot was performed.We found that only DNA extracted from cells treated with compound 5 and irradiated at 365 nm could be developed (channel 4 in Fig. 5), which indicating that genomic DNA was indeed modified with alkyne which was caused by EdU released from decaging compound 5.

|
Download:
|
| Fig. 5. Analyze genomic DNA modified with alkyne groups by dot blot. The HeLa cells were treated with PBS, EDU, 5 or 5+UV, and the genomic DNA was extracted and subjected to dot blot, corresponding to 1, 2, 3 and 4 channels, respectively. Channels 2 and 4 revealed positive, and Channels 1 and 3 were negative. | |
{kind=link}
In conclusion, we attached a photocage group to possible coupling site within EdU, the N-3 position of the base and the 3' OH groups of the deoxyribose In vitro, we found that both of them have good photolysis efficiency. In the subsequent cell experiments, using bioorthogonal chemistry, it was found that the derivative linked by methylene at the N-3 position of EdU was not involved in metabolism, and desired genomic DNA modified with alkyne could be obtained by photodecaging of it. The carbonate-linked derivative at the 3' oxygen atom has a low dissociation rate, thus, it can participate in metabolism in vivo without photodecaging. In this research, the complex process of cell metabolism is regulated by optical channel, which can provide a reference for evaluating whether small molecules are involved in metabolism after photocaging modification. It is more desirable to use alkyl linkages instead of carbonyls when caging some bioactive molecules such as drugs and inhibitors [25, 26]. This is more conducive to biochemists designing more reasonable caging strategies for controlling whole cells at a high spatial and temporal resolution.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsThis work was supported by the National Natural Science Foundation of China (Nos. 21432008, 91753201 and 21721005). We thank the large-scale instrument and equipment sharing foundation of Wuhan University.
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.10.021.
| [1] |
P. Klan, T. Šolomek, C.G. Bochet, et al., Chem. Rev. 113 (2013) 119-191. DOI:10.1021/cr300177k |
| [2] |
G.C.R. Ellisdavies, Nat. Methods 4 (2007) 619-628. DOI:10.1038/nmeth1072 |
| [3] |
A. Deiters, ChemBioChem 11 (2010) 47-53. |
| [4] |
J. Hemphill, E.K. Borchardt, K. Brown, A. Asokan, A. Deiters, J. Am. Chem. Soc. 137 (2015) 5642-5645. DOI:10.1021/ja512664v |
| [5] |
H. James, C. Chungjung, J.W. Chin, D. Alexander, J. Am. Chem. Soc. 135 (2013) 13433-13439. DOI:10.1021/ja4051026 |
| [6] |
L. Lele, T. Rong, C. Hunghao, et al., Proc. Natl. Acad. Sci. U. S. A. 111 (2014) 17099-17103. DOI:10.1073/pnas.1420105111 |
| [7] |
H. Alexander, M. Günter, J. Am. Chem. Soc. 127 (2005) 822-823. DOI:10.1021/ja043285e |
| [8] |
D.D. Young, M.O. Lively, D. Alexander, J. Am. Chem. Soc. 132 (2010) 6183-6193. DOI:10.1021/ja100710j |
| [9] |
J.M. Govan, M.O. Lively, D. Alexander, J. Am. Chem. Soc. 133 (2011) 13176-13182. DOI:10.1021/ja204980v |
| [10] |
H. Wang, R. Wang, K. Cai, et al., Nat. Chem. Biol. 13 (2017) 415-424. DOI:10.1038/nchembio.2297 |
| [11] |
F. Li, Y. Zhang, J. Bai, et al., J. Am. Chem. Soc. 139 (2017) 10617-10620. |
| [12] |
N. Ha Phuong, S. Stewart, M.N. Kukwikila, et al., Angew. Chem. Int. Ed. 58 (2019) 6620-6624. DOI:10.1002/anie.201901139 |
| [13] |
L. Jie, R.C. Peng, Nat. Chem. Biol. 12 (2016) 129-137. DOI:10.1038/nchembio.2024 |
| [14] |
S. Adrian, T.J. Mitchison, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 2415-2420. DOI:10.1073/pnas.0712168105 |
| [15] |
H. Lusic, A. Deiters, Synthesis 13 (2006) 2147-2150. |
| [16] |
L. Hrvoje, D.D. Young, M.O. Lively, D. Alexander, Org. Lett. 9 (2007) 1903-1906. DOI:10.1021/ol070455u |
| [17] |
C. Feng, Y. Li, R.C. Spitale, Org. Biomol. Chem. 15 (2017) 5117-5120. DOI:10.1039/C7OB01009E |
| [18] |
U. Kosinska, C. Carnrot, S. Eriksson, L. Wang, H. Eklund, FEBS J. 272 (2010) 6365-6372. |
| [19] |
H. Lusic, R. Uprety, A. Deiters, Org. Lett. 12 (2010) 916-919. DOI:10.1021/ol902807q |
| [20] |
J. Kapuscinski, Biotech. Histochem. 70 (1995) 220-233. DOI:10.3109/10520299509108199 |
| [21] |
F.A. Tanious, J.M. Veal, H. Buczak, L.S. Ratmeyer, W.D. Wilson, Biochemistry 31 (1992) 3103-3112. DOI:10.1021/bi00127a010 |
| [22] |
H. Haizheng, W. Yinsheng, Anal. Chem. 79 (2007) 322-326. DOI:10.1021/ac061465w |
| [23] |
F.T. Liu, N.J. Leonard, J. Am. Chem. Soc. 101 (1979) 996-1005. DOI:10.1021/ja00498a034 |
| [24] |
M. Wilchek, E.A. Bayer, Immunol. Today 5 (1984) 39-43. DOI:10.1016/0167-5699(84)90027-6 |
| [25] |
K. Neumann, A. Gambardella, A. Lilienkampf, M. Bradley, Chem. Sci. 9 (2018) 7198-7203. DOI:10.1039/C8SC02610F |
| [26] |
J. Tu, M. Xu, S. Parvez, R.T. Peterson, R.M. Franzini, J. Am. Chem. Soc. 140 (2018) 8410-8414. DOI:10.1021/jacs.8b05093 |