 2020, Vol. 31
2020, Vol. 31 


b The First Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China
Genetic variation, which is estimated to be 0.1%–0.5% at nucleic acid level, is a crucial factor for the occurrence of many diseases, especially cancers [1, 2]. The detection of tumor-associated site-specific genetic mutations in circulating blood has become an important and promising way for cancer diagnosis at the early stages. Notably, DNA mutation canbe transcribed into mRNA variation, which may directly alter the structure or function of the corresponding protein. Therefore, although DNA mutation analysis has been intensively investigated [3, 4], in some cases, the detection of cellfree circulating mutant mRNA, rather than DNA mutation, is more intuitive and valuable for liquid biopsy of cancers [5]. On the other hand, the initial mRNA mutation-driven tumorigenesis usually present in a handful number of infrequent cells in tissues or organisms and the level of mutant mRNA between cancer cells and normal cells may vary greatly, so sensitive and accurate detection of mutant mRNA in a single cell is also of great interest for cancer research [6-9]. A desirable method that enables the accurate detection of mRNA mutation in liquid biopsy or in single cells should possess both high sensitivity and high specificity, since the coexistent of a large number of normal mRNA may mask the clues of rare cancer-associated mutant mRNA.
Although some enzyme-free methods have been proposed to detect RNA or RNA nucleotide variations [10-14], up to now, the most widely used method for detecting mRNA mutation is allelespecific reverse transcription (RT)-PCR [15, 16]. RNA is converted to cDNA by reverse transcription, and the mutation site on the cDNA is detected by subsequent PCR. The discrimination of the sitespecific mutation depends on the ability of polymerase to identify the mismatch between the cDNA and the 3' end of the primer, so the specificity is limited and it is generally difficult to detect mutation frequencies of less than 1% [17]. Since a large number of wild sequences may coexist with the mutants, allele-specific RTPCR cannot high-specifically detect mRNA mutation, particularly, at the single-cell level or in liquid biopsy. In addition, it is not always easy to design perfect primer pairs for PCR at the mutation site, wherefore application of allele-specific RT-PCR is also hindered by the target sequence. As an alternative, ligase chain reaction (LCR) is also applied to the detection of mRNA as well as its mutation by using ligase to discriminate the base variation, which also suffers from the low specificity [18]. Therefore, it is still imperative to establish highly specific and sensitive methods for the accurate quantitation of site-specific mutant mRNA in the presence of abundant normal mRNA. For an ideal protocol, the interference of wild-type mRNA should be completely eliminated.
Herein, we have elegantly designed an LNA (locked nucleic acid)-modified probe which can selectively guide the RNase H to cleave only the wild-type mRNA (wtRNA) while the mutant mRNA (mutRNA) will remain intact. The intact mutRNA can be amplified and detected by real-time RT-PCR but the disconnected wtRNA will be not replicated at all by PCR. Based on the highly selective depletion of the wtRNA, the accuracy for detection of rare RNA mutation has been greatly boosted.
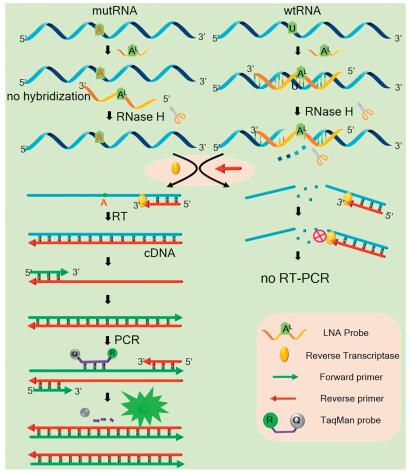
In this study, the mutant BRAF mRNA fragment with the V600E mutation site (see details in Table S1 in Supporting infromation) is used as a model target where the base U of the wtRNA is converted to A in the mutRNA. This V600E site mutation has close correlation with the occurrence of melanoma [19, 20]. The mechanism of the LNA probe-assisted RT-PCR assay is schematically illustrated in Fig. 1. An 11-nt DNA probe (defined as LNA probe), which contains an LNA nucleotide (AL) exactly opposite to the mutant site, is fully complementary to the wtRNA. The introduction of an LNA nucleotide can obviously increase the melting temperature (Tm) of the formed LNA probe/wtRNA duplexes [21]. As a result, the Tm of the LNA probe/wtRNA duplex is much higher than that of the LNA probe/mutRNA duplex because the AL nucleotide in the LNA probe is mismatched with the mutated A nucleotide (Fig. S1 in Supporting information).

|
Download:
|
| Fig. 1. Schematic illustration of the LNA probe-assisted RT-PCR for mRNA mutation detection. | |
{kind=link}
Therefore, in a wide temperature range, the LNA probe can only stably bind to wtRNA, but will not hybridize with mutRNA. RNase H is an endoribonuclease which can selectively hydrolyze the RNA strand in the RNA/DNA heteroduplexes [22, 23]. Under proper temperature, wtRNA perfectly pairs with the LNA probe, and thus will be completely digested by RNase H at the hybridizing site. Accordingly, subsequent RT-PCR cannot be performed, which will no longer interfere with mutRNA detection. Meanwhile, due to the AL/A single-base mismatch, the Tm of LNA probe/mutRNA duplex is much lower. As such, the LNA probe cannot hybridize with the mutRNA, and the RNase H-based digestion reaction cannot occur on the mutRNA at all. So, the RT-PCR amplification of the mutRNA can be proceeded normally, during which the TaqMan probe is hydrolyzed by Taq Hot Start DNA polymerase to generate fluorescent signal.
This design has several unique advantages. In principle, compared with allele-specific PCR, the primer does not need to be exactly designed at the mutation site, making the primer design more flexible and reliable. Therefore, this method can be applied to the quantification of single-base mutations in arbitrary RNA sequences at any site. Most importantly, the high background of wtRNA can be thoroughly depleted with high specificity while the mutRNA will be not affected, which can guarantee the accurate and reliable detection of rare mutRNA in real biomedical samples without interference of wtRNA. The high specificity of this strategy should be mainly attributed to the LNA probe-elevated difference of Tm values between LNA probe/wtRNA and LNA probe/mutRNA, which allows RNase H to effectively and selectively deplete the wtRNA over a wide digestion temperature range (37-42 ℃, Fig. S1).
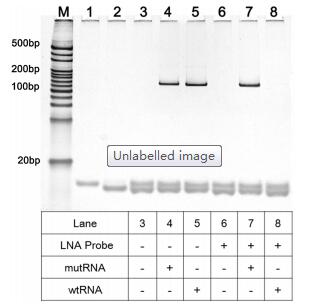
The crucial role of LNA probe in distinguishing mRNA mutation is verified by polyacrylamide gel electrophoresis (PAGE). As depicted in Fig. 2, one can see that if the LNA probe is absent, the clear band of PCR products for wtRNA (128 bp) can be identified (lane 5). However, when the LNA probe is introduced into the digestion reaction, such PCR product band for wtRNA can no longer be observed (lane 8), indicating that the LNA probe can efficiently hybridize with the wtRNA to make the wtRNA be completely depleted by RNase H. In contrast, for the mutRNA, the bands of the RT-PCR product can be clearly seen with almost the same pixel intensities either in the absence (lane 4) or presence (lane 7) of LNA probe (~128 bp), suggesting that LNA probe can only selectively guide the depletion of wtRNA by RNase H while the mutRNAtemplated RT-PCR will not be influenced. All these results prove that the proposed LNA probe-assisted approach exhibits excellent specificity in discriminating site-specific single-base mutation of mRNA. In virtue of this, the nonspecific interference of normal RNA can be effectively eliminated in real samples, and thus accurate quantification of mutRNA can be acquired.

|
Download:
|
| Fig. 2. Non-denaturing polyacrylamide gel electrophoresis results of wtRNA or mutRNA in different experimental conditions. Lane 1: forward primer; Lane 2: reverse primer; Lane 3: forward primer/reverse primer; Lanes 4-8 show the RT-PCR products for mutRNA or wtRNA after cleaved by RNase H with presence or absence of LNA-probe, with the RNase H reaction temperature of 42 ℃. Both the concentrations of wtRNA and mutRNA are 1 fmol/L. | |
{kind=link}
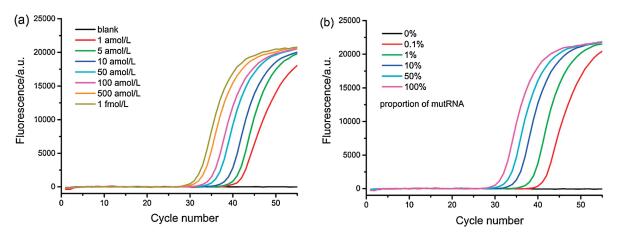
Some important experimental parameters including the temperature of the RNase H digestion reaction, the concentration of LNA probe and the amount of RNase H, which may influence the performance for mutRNA analysis, are all optimized (Figs. S1–S3 in Supporting information). Ultimately, 42 ℃ for RNase H digestion, 12 U RNase H and 1 μmol/L LNA probe are found to be the optimal. Under such conditions, series dilutions of mutRNA from 1 amol/L to 1 fmol/L were detected via the proposed LNA probe-assisted RT-PCR assay, and the well-shaped real-time fluorescence curves were exhibited in Fig. 3a. It is evident that the fluorescence curves arise more rapidly with the presence of higher concentrations of mutRNA, leading to the gradual reduction of the corresponding Ct value. Significantly, the fluorescence response generated by 1 amol/L mutRNA, 6 copies in a 10 μL volume, can be obviously distinguished from that of blank control, indicating that the proposed method possesses an ultrahigh sensitivity to detect mutRNA at the single-molecule level. The Ct values are linearly proportional to the logarithm (lg) of mutRNA concentrations ranging from 1 amol/L to 1 fmol/L (Fig. S4 in Supporting information). The linear regression equation is Ct = -18.81 – 3.45lgCmutRNA(mol/L) with a correlation coefficient (R2) of 0.9942.

|
Download:
|
| Fig. 3. (a) Real-time fluorescence curves generated by mutRNA with different concentrations. From right to left, the mutRNA concentrations are successively 0 (blank), 1 amol/L, 5 amol/L, 10 amol/L, 50 amol/L, 100 amol/L, 500 amol/L, and 1 fmol/L, respectively. (b) The real-time fluorescence curves aroused by the mixture of wtRNA/mutRNA with different proportions (the concentration of total RNA is fixed at 1 fmol/L). From right to left, the proportion of mutRNA in the RNA mixture is 0% (without mutRNA), 0.1% (1 amol/L mutRNA), 1% (10 amol/L mutRNA), 10% (100 amol/L mutRNA), 50% (500 amol/L mutRNA) and 100% (1 fmol/L mutRNA), respectively. | |
{kind=link}
Furthermore, mutRNA and wtRNA are mixed at different ratios of 0-100% with constant total RNA of 1 fmol/L. Such mixtures directly act as samples to evaluate the specificity of the proposed mutRNA assay. As exhibited in Fig. 3b, no exponential fluorescence curve can be aroused by 1 fmol/L wtRNA (0% of mutRNA), the response of which completely overlaps with the blank. Nevertheless, as the ratio of mutRNA increases in the mixture, the fluorescence curve arises more rapidly. As low as 0.1% mutRNA (equal to 6 molecules) can be effectively distinguished in the presence of vast number of wtRNA (1 fmol/L, equal to 6000 molecules), indicating a high specificity of the proposed strategy for the selective quantification of single-base mutated RNA at the single-molecule level. Moreover, the Ct values respectively produced by 0.1% (1 amol/L), 1% (10 amol/L), 10% (100 amol/L), and 100% (1 fmol/L) mutRNA in the RNA mixtures are all consistent with those produced by the same concentrations of pure mutRNA, indicating that the positive amplification signals are only generated by the mutRNA in the mixtures. Such results indicate that the potential false positive signals from wild-type sequences can be completely precluded by using the LNA probe-assisted digestion strategy, which can ensure the accurate quantification of rare mutRNA in complex biological samples.
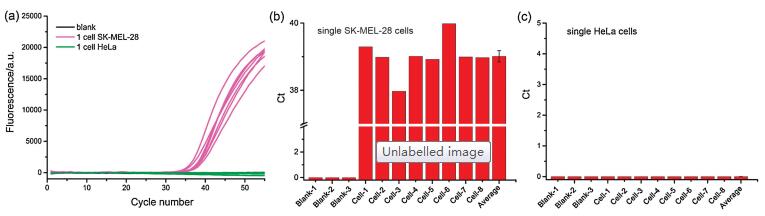
Traditional mRNA analysis in a large population of cells may mask the important information of mRNA fluctuation in individual single cells. Therefore, it is of particular significance to accurately quantify the amount of mutRNA in single cells, which can further elucidate the heterogeneity and complexity of cancer development. In this work, we have further applied the LNA probe-assisted RT-PCR method to the detection of mutRNA in individual single cells. Previous literature has suggested that the target site of BRAF mRNA in SK-MEL-28 cells is homozygous mutant type, while that in HeLa cells is homozygous wild type [24, 25], which is consistent with the sequencing results from HeLa cells and SK-MEL-28 cells, respectively (Fig. S5 in Supporting information). The single cells are individually captured with a micromanipulator system. After lysis at 95 ℃ for several minutes, the amount of mutRNA in each lysate of single cell is separately determined with the established RT-PCR approach. One can see from Figs. 4a and b that the positive exponential amplification signals produced by 8 randomly obtained single SK-MEL-28 cells can be all obviously identified. The determined copy numbers of mutRNA in the eight individual SK-MEL-28 cells varies from 51 to 197, due to the stochastic nature of gene expression in single cells. On average, the calculated copy number of mutRNA in the detected SK-MEL-28 cells is 104/cell, consisting with the values obtained from the lysate of 10 (~132 copies/cell) or 100 SK-MEL-28 cells (~102 copies/cell), respectively. As mentioned above, there is no BRAF V600E mutRNA in HeLa cells, so no fluorescent response is generated by all of the single HeLa cells in Fig. 4c. All these results demonstrate that due to its ultrahigh specificity and sensitivity, the LNA probe-assisted RT-PCR strategy may serve as a reliable and promising tool for the detection of mutRNA in individual single cells.

|
Download:
|
| Fig. 4. Quantitative detection of mutRNA in single cells. (a) The real-time fluorescence amplification curves generated by individual single SK-MEL-28 cells (purple lines), single HeLa cells (green) and blank control (black); (b) The corresponding Ct values produced by the single SK-MEL-28 cells; (c) The corresponding Ct values produced by the single HeLa cells. | |
{kind=link}
In conclusion, by integrating the elegant LNA probe-assisted selective depletion of wtRNA, and high amplification efficiency of RT-PCR, we have proposed a flexible and practical approach to detect single-base mutated mRNA with ultrahigh selectivity. The mutRNA-specific assay is realized with the help of selective RNase H digestion, which can completely disconnect wtRNA without affecting the mutRNA. Based on this advantage, the interference of wtRNA can be favourably eliminated in complex biological samples. Besides for the high specificity, this LNA probe-assisted RT-PCR assay also achieves an ultrahigh sensitivity, which enables the accurate quantification of mutRNA target at single molecule and single cell level. This method can be universally applicable to detecting any RNA mutation site by specifically designing the site-specific LNA probe. It should be noted that for the detection of different mutation sites of interest, the site-specific LNA probes as well as the RNase H cleavage temperatures should be optimized case by case. In virtue of its easy design, wide temperature window, high sensitivity and specificity, the established LNA probe-assisted RT-PCR strategy provides a robust platform for the study of the function of mutRNA at the single cell level and for the mutRNA-associated liquid biopsy.
AcknowledgmentsThis work is supported by the National Natural Science Foundation of China (Nos. 21622507 and 21575086), the Program for Changjiang Scholars and Innovative Re-search Team in University (No. IRT_15R43) and the Fundamental Research Funds for the Central Universities (No. GK201802016).
Appendix A. Supplementary dataSupplementary material related to this article can befound, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.015.
| [1] |
M.G. Mohsen, D.B. Ji, E.T. Kool, Chem. Sci. 10 (2019) 3264-3270. DOI:10.1039/C8SC03901A |
| [2] |
S. Levy, G. Sutton, P.C. Ng, et al., PLoS Biol. 5 (2007) e254.. DOI:10.1371/journal.pbio.0050254 |
| [3] |
J.B. Long, Y.X. Liu, Q.F. Cao, et al., Chin. Chem. Lett. 26 (2015) 1031-1035. DOI:10.1016/j.cclet.2015.05.036 |
| [4] |
D.B. Zhu, D. Xing, Y.B. Tang, Chin. Chem. Lett. 18 (2007) 869-871. DOI:10.1016/j.cclet.2007.05.002 |
| [5] |
J. Das, I. Ivanov, L. Montermini, et al., Nat. Chem. 7 (2015) 569-575. DOI:10.1038/nchem.2270 |
| [6] |
C.E. Meacham, S.J. Morrison, Nature 501 (2013) 328-337. DOI:10.1038/nature12624 |
| [7] |
C.H. Zong, S.J. Lu, A.R. Chapman, et al., Science 338 (2012) 1622-1626. DOI:10.1126/science.1229164 |
| [8] |
A. Rakszewska, R.J. Stolper, A.B. Kolasa, et al., Angew. Chem. Int. Ed. 55 (2016) 6698-6701. DOI:10.1002/anie.201601969 |
| [9] |
C. Larsson, I. Grundberg, O. Söderberg, et al., Nat. Methods 7 (2010) 395-397. DOI:10.1038/nmeth.1448 |
| [10] |
S.Z. Yue, X.Y. Song, W.L. Song, et al., Chem. Sci. 10 (2019) 1651-1658. DOI:10.1039/C8SC04756A |
| [11] |
Y.F. Li, S.Z. Yue, H.J. Qi, et al., Chem. Commun. 55 (2019) 4103-4106. DOI:10.1039/C9CC00747D |
| [12] |
S. Bi, S.Z. Yue, S.S. Zhang, Chem. Soc. Rev. 46 (2017) 4281-4298. DOI:10.1039/C7CS00055C |
| [13] |
X.D. Li, B. Chen, L. Lan, et al., Chin. Chem. Lett. 29 (2018) 1637-1640. DOI:10.1016/j.cclet.2018.06.003 |
| [14] |
C.B. Qi, H.P. Jiang, J. Xiong, et al., Chin. Chem. Lett. 30 (2019) 553-557. DOI:10.1016/j.cclet.2018.11.029 |
| [15] |
J. Morlan, J. Baker, D. Sinicropi, PLoS One 4 (2009) e4584.. DOI:10.1371/journal.pone.0004584 |
| [16] |
G.M. Arechavaleta, V. Scholl, V. Pérez, et al., Clin. Exp. Med. 11 (2011) 55-59. DOI:10.1007/s10238-010-0101-x |
| [17] |
W.W. Chen, L. Balaj, L.M. Liau, et al., Mol. Ther.-Nucl. Acids 2 (2013) e109. DOI:10.1038/mtna.2013.28 |
| [18] |
Y.L. Hu, H.X. Jia, Y.C. Wang, et al., Chem. Commun. 50 (2014) 13093-13095. DOI:10.1039/C4CC05102E |
| [19] |
D.J. Panka, R.J. Sullivan, J.W. Mier, Melanoma Res. 20 (2010) 401-407. |
| [20] |
D. Dankort, D.P. Curley, R.A. Cartlidge, et al., Nat. Genet. 41 (2009) 544-552. DOI:10.1038/ng.356 |
| [21] |
Y. You, B.G. Moreira, M.A. Behlke, et al., Nucleic Acids Res 34 (2006) e60. DOI:10.1093/nar/gkl175 |
| [22] |
C.Y. Lee, H. Jang, K.S. Park, et al., Nanoscale 9 (2017) 16149-16153. DOI:10.1039/C7NR04060A |
| [23] |
M. Nowotny, S.A. Gaidamakov, R.J. Crouch, et al., Cell 121 (2005) 1005-1016. DOI:10.1016/j.cell.2005.04.024 |
| [24] |
B. Sapkota, C.E. Hill, B.P. Pollack, OncoImmunology 2 (2013) e22890. DOI:10.4161/onci.22890 |
| [25] |
A. Nakamura, T. Arita, S. Tsuchiya, et al., Cancer Res. 73 (2013) 7043-7055. DOI:10.1158/0008-5472.CAN-13-1825 |