 2020, Vol. 31
2020, Vol. 31 


Rhenium is able to have a variety of valences from zero to 7+ and this property can be connected to multifunctionality of Re catalysts. Metallic Re species has high activation ability of H2 molecule, and Re with higher valence can have higher acidity, and so on. In addition, it is characteristic that Re has higher oxophilicity than other noble metals such as Pt, Ir. Possible multivalence property of Re can make the valence control difficult. Conversely, the control of the valence enables the extraction of functions on Re catalysts. A variety of Re catalysts has been recently applied to the reactions for the production of biomass-derived fuels and chemicals as mentioned in the reviews [1-10]. It has been known that the oxygen content of biomass-derived substrates (such as cellulose or sugars) is much higher than the value-added chemicals (such as a monomer for the production of plastics, resins). Building blocks in biomass refineries have been proposed and they also have high oxygen content [11]. Therefore, the reactions to decrease the oxygen contents become more and more important. The reactions include dehydration, reduction, and so on. In particular, reductants are necessary for the reduction and H2 is one of the green reductants in terms of atom economy.Hydrodeoxygenation, C-O hydrogenolysis, deoxydehydration of biomass-related substrates with H2reductant canbeincluded in the reduction. Here, the development of heterogeneous catalysts can increase the feasibility of the target process because of easy separationof the catalystsfrom the products and catalyst reusability. This short review introduces the development of heterogeneous Re catalysts for deoxydehydration of vicinal OH groups with H2 reductant using high valence Re species, C-O hydrogenolysis using the combination of low valence Re species with noble metals, and hydrogenation of carboxylic acids using the combination of metallic Re with cationic Re species.
2. Highvalence Re species on oxide supports for deoxydehydration of vicinal OH groups with H2 reductantDeoxydehydration is the removal of vicinal OH groups to give carbon-carbon double bond as described below (Eq. 1).
|
(1) |
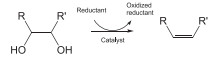
It has been reported that homogeneous Re(Ⅶ) complex such as C5Me5ReO3 and CH3ReO3 catalyzes the deoxydehydration using PPh3 and secondary alcohols as a reductant [12,13]. These Re complexes have low ability to activate H2, and H2 is not suitable reductant for the deoxydehydration. It has been proposed that the catalytic cycle consists of the reduction of Re(Ⅶ) to Re(Ⅴ) with the 2-electron reductant (PPh3 or secondary alcohols) and the successive oxidation of Re(Ⅴ) in the Re diolate to give Re(Ⅶ) and the product with carbon-carbon double bond, which is regarded as the redox mechanism between Re(Ⅶ) and Re(Ⅴ) as illustrated in Fig. 1.

|
Download:
|
| Fig. 1. Catalytic cycle of deoxydehydration of the substrates with vicinal OH groups using CH3ReO3 and 2-electron reductant. | |
{kind=link}
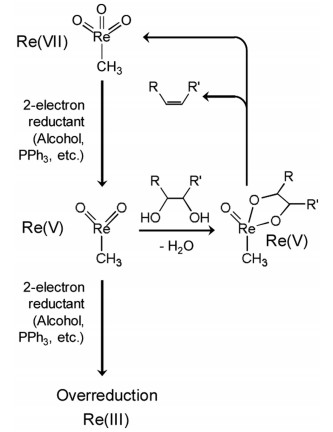
There has been a previous report on the deoxydehydration of styrene glycol, 1, 2-tetradecanediol, and (+)-diethyl tartrate with H2 reductant to their products with carbon-carbon double bond in moderate-to-excellent yield over ReOx/C [14]. The activity per Re amount on ReOx/C in the deoxydehydration with H2 reductant was clearly lower than that of homogeneous CH3ReO3 catalyst in deoxydehydration with 3-pentanol reductant. Our group has been developing more active heterogeneous Re catalysts for the deoxydehydration. Our approach for the catalyst development is the combination of high valent Re species fixed on oxides and noble metals with high H2 activation ability, and we found that CeO2 was much more effective support than carbon, SiO2, ZrO2, TiO2, MgO, La2O3, and Al2O3 [15]. Regarding CeO2-supported ReOx catalyst (ReOx/CeO2), the modification effect of additive metals was investigated, and it is found that the Pd-added ReOx/CeO2 (ReOx-Pd/CeO2) showed high activity and selectivity to saturated product in the deoxydehydration + hydrogenation of 1, 4-anhydroerythritol to tetrahydrofuran. The formation of saturated product can be due to high catalytic activity of Pd particles in hydrogenation of carbon-carbon double bond. The catalytic activity per Re amount of ReOx-Pd/CeO2 was clearly higher than that on CH3ReO3 in the deoxydehydration of 1, 4-anhydroerythritol to 2, 5-dihydrofuran [15,16]. An interesting behavior of ReOx-Pd/CeO2 is the effect of loading amount of Re. In the range of 0.5–2.0 wt% Re, the activity per g-cat increased with the increasing loading amount of Re. In the range of 2.0–10 wt%, the activity decreased with the decreasing loading amount of Re [15,16]. The optimum loading amount of Reis 2.0 wt%.The effect of loading amount on the catalytic activity and catalyst characterization indicates that monomeric Re species on CeO2 is a catalytically active site in the deoxydehydration [16]. Only Re4+ and Re6+ species were detected in the XPS of highly active ReOx-Pd/CeO2 (2 wt% Re, 0.3 wt% Pd) after the reaction (Fig. 2), suggesting that the deoxydehydration proceeds by the redoxof Re6+ and Re4+. In contrast, in the XPS of ReOx-Pd/SiO2 (2 wt% Re, 0.3 wt% Pd) after the reaction, the presence of Re4+ and Re0 species was verified (Fig. 2) [16]. It is clear that Re species tend to be reduced more deeply on ReOx-Pd/SiO2, which can be connected to the low catalytic activity due to overreduction of Re species. At the same time, the comparison of the results of CeO2 and SiO2 indicates that CeO2 play a crucial role on the suppression of overreduction of Re species. In addition, in order to estimate the number of active site (monomeric Re species), the stoichiometric deoxydehydration of 1, 2-hexanediol on the pre-reduced ReOx-Pd/CeO2 with various loading amounts of Re was carried out [16]. As a result, it is found that the ratio of the number of active Re species to total Re amount decreases with increasing the loading amount of Re. In the case of ReOx-Pd/CeO2 (2 wt% Re, 0.3 wt% Pd), the ratio is estimated to be about 0.4, meaning that almost half of Re species can be catalytically active site [16].

|
Download:
|
| Fig. 2. XPS of reacted catalysts in Re 4f region. (a) ReOx-Pd/CeO2 after reaction (2 wt% Re, 0.3 wt% Pd, reaction temperature = 413 K, t = 4 h), (b) ReOx-Pd/SiO2 after reaction (2 wt% Re, 0.3 wt% Pd, reaction temperature = 413 K, t = 4 h). Raw XPS data (black solid line), calculated data (black dotted line). Reference: C 1s = 284.6 eV. Reprinted from with permission [16]. Copyright 2016, American Chemical Society. | |
{kind=link}
Another important behavior is the kinetics of deoxydehydration + hydrogenation of 1, 4-anhydroerythritol to tetrahydrofuran. ReOx-Pd/CeO2 (2 wt% Re, 0.3 wt% Pd) showed almost zero reaction order with respect to H2 pressure, indicating that the deoxydehydration of the Re-diolate species [16], which can be an intermediate from the studies on the homogeneous Re complexes, is the rate-determining step. The rate of the elementary steps including H2 activation and the reduction of Re species, which can be strongly promoted by Pd, is much higher than that of deoxydehydration step. Pd has high H2 activation ability to promote the reduction of Re species, at the same time, CeO2 has the property for the strong suppression of the overreduction of Re species. Combination of the role of Pd and CeO2 enables high performance of ReOx-Pd/CeO2.
A problem of ReOx-Pd/CeO2 catalyst is that unsaturated products in the deoxydehydration are not obtained. The key is the component to activate H2 to promote the reduction of Re species on CeO2 without the ability to hydrogenate carbon-carbon double bond in the unsaturated deoxydehydration product. As a result of metal survey as additive to ReOx/CeO2, we found that ReOx-Au/CeO2 gave unsaturated products such as allyl alcohol, 2, 5-dihydrofuran and 1, 3-butadiene in the deoxydehydration of glycerol, 1, 4-anhydroerythritol, and erythritol with H2 reductant, respectively [17,18]. Since Au nanoparticle with smaller size has higher catalytic activity, rather large Au particles (12 nm) exhibited high selectivity and high yield of unsaturated products, and the ReOx-Au/CeO2 catalysts are regarded as truly heterogeneous catalysts for the deoxydehydration with H2 reductant [17,18]. The particle size of CeO2 on ReOx-Au/CeO2 was estimated to be about 8 nm, which is almost comparable to the size of Au (12 nm). In contrast, Pd metal particles on ReOx-Pd/CeO2 are highly dispersed. In the case of ReOx-Au/CeO2 (2 wt% Re, 0.3 wt% Au, particle size of Au = 12 nm), the number ratio of Au particles to CeO2 particles is calculated to be 1:3000 [18]. The number of Au particles is very small compared to that of CeO2 and Re species on CeO2 (Fig. 3a), but the role of Au particles covers almost all the Re species on CeO2, which can be explained by the spillover phenomenon of the hydrogen species formed on Au surface or Au-CeO2 interface (Fig. 3b) [18].

|
Download:
|
| Fig. 3. Illustration of each component of ReOx-Au/CeO2 (2 wt% Re, 0.3 wt% Au, particle size of Au: 12 nm, size of CeO2: 8 nm) (a), and the model scheme of H2 activation and spillover from Au to ReOx via CeO2 (b). Reprinted from with permission [18]. Copyright 2018, American Chemical Society. | |
{kind=link}
This mechanism is also the case of ReOx-Pd/CeO2 catalysts, where hydrogen species can be supplied to Re species more rapidly than the case of ReOx-Au/CeO2. This can be because Pd metal particles have much higher dispersion than the case of Au and Pd has usually higher activity of H2 activation than Au.
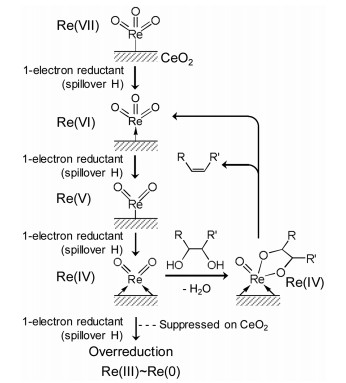
Here, the difference in the valences of the active Re species on CeO2 and homogeneous Re complex such as CH3ReO3 is discussed. As shown in Fig. 1, CH3ReO3 has the two states of Re7+ and Re5+ during the deoxydehydration because 2-election reductant (secondary alcohol) and 2-electron oxidant (substrate with vicinal OH groups) are used for the catalytic reaction. In contrast, when H2 is used as a reductant and the dissociated hydrogen species can be a real reductant on ReOx-M/CeO2 catalyst, the valence of Re species can be changed step by step as shown in Fig. 4 [18]. An important point is that the overreduction of Re species from Re4+ can be suppressed by the presence of CeO2 support. This mechanism is unclear at present, however, further investigation such as DFT calculation is necessary [19].

|
Download:
|
| Fig. 4. Catalytic cycle of deoxydehydration of the substrates with vicinal OH groups using ReOx-M/CeO2 (M = Pd, Au) and H2 reductant. | |
{kind=link}
Fig. 5 shows the applicability of ReOx-Au/CeO2 for deoxydehydration and ReOx-Pd/CeO2 for deoxydehydration + hydrogenation using H2 reductant, which can contribute to the production of biomass-derived chemicals [15-18,20-23].

|
Download:
|
| Fig. 5. Example of application of ReOx-Au/CeO2 for deoxydehydration and ReOx-Pd/CeO2 for deoxydehydration + hydrogenation using H2 reductant. | |
{kind=link}
3. Low valent Re species on metal surface for C–O hydrogenolysis
Our group reported that Rh-ReOx/SiO2 showed much higher catalytic activity in the C–O hydrogenolysis of glycerol in 2009, although the selectivity to 1, 3-propanediol was not high [24]. Effective combination of noble metals with ReOx is a little limited, and Rh-ReOx and Ir-ReOx showed high catalytic performance in the C–O hydrogenolysis [25-47]. The chemical state of Re species on noble-metal-ReOx based catalysts has been investigated. The structural change of Rh-ReOx/SiO2 (4 wt% Rh, Re/Rh = 0.5) during the temperature programmed reduction (TPR) with H2 was investigated using in situ Re L3-edge and Rh K-edge quick-scanning X-ray absorption fine structure in the temperature range of room temperature to 600 K [31]. Rh-ReOx/SiO2 (4 wt% Rh, Re/Rh = 0.5) showed high catalytic activity and selectivity in the C–O hydrogenolysis of tetrahydrofurfuryl alcohol to 1, 5-pentanediol [25]. The valence of Rh and Re on Rh-ReOx/SiO2 (Re/Rh = 0.5) was determined individually from the coordination number of Rh–O bond in the Rh K-edge EXAFS analysis and the chemical shift of the binding energy in the Re L3-edge XANES analysis. It is concluded that the reduction of Re species followed that of Rh, although Rh-ReOx/SiO2 gave single peak in the TPR profile [31]. An interesting point is that the average valence of Re is maintained to be around 2+ in the temperature range of 373–600 K, and the further reduction of Re cation to metallic Re can be suppressed, although the reduction of Re is strongly promoted by the presence of Rh. At the same time, the Re L3-edge EXAFS analysis indicates that the Re-O, Re-Rh, and Re-Re bonds are detected on Rh-ReOx/SiO2 (Re/Rh = 0.5) [31]. In particular, the bond length of Re-Rh and Re-Re bonds is about 0.265 nm and 0.270 nm, respectively. These bond lengths are very close to the sum of metal bond radius of Rh and Re, indicating the direct bond between Re atom and Rh atom. Based on the average valence of Re (~2+), partially oxidized Re species can be located on the surface of Rh metal surface [31]. Considering the coordination numbers of Re-Rh, Re-Re and Re-O bonds, 2-dimensional clusters of Re oxides in low valence can be formed on the surface on Rh metal particles [31].
The structure of Pt-ReOx/SiO2 (2 wt% Pt, Re/Pt = 0.2 and 0.5) was alsoexaminedby X-rayabsorption spectroscopy [48].TheRe L3-edge EXAFS analysis indicates that the Re-O, Re-Pt, and Re-Re bonds are detected on Pt-ReOx/SiO2 catalysts. The bond length of Re-Pt and Re-Re bonds is about 0.275 nm and 0.268 nm, respectively. These bondlengths areveryclose to the sum of metal bond radius of Re and Pt, indicating the direct bond between Re atom and Pt atom. Based on the average valence of Re (~3+), partially oxidized Re species can be located on the surface of Pt metal surface. Considering the coordination numbers of Re-Pt, Re-Re and Re-O bonds, 2-dimensional clusters of Re oxides in low valence can be also formed on the surface on Pt metal particles [48]. As a result, the structure of ReOx on Rh and Pt metal surface can be similar. On the other hand, the catalytic activity of Rh-ReOx/SiO2 and Pt-ReOx/SiO2 in the C-O hydrogenolysis of glycerol and tetrahydrofurfuryl alcohol was very different regardless of similar structure [26]. According to the kinetics analysis of the C-O hydrogenolysisof tetrahydrofurfuryl alcohol to 1, 5-pentanediol, the reaction order with respect to H2 pressure is first, indicating that the reaction of hydrogen species can be rate-determining step [29]. The activity difference may be explained by the ability to form the active hydrogen species such as hydride, however, further investigation is necessary for the explanation supported by DFT studies and so on.
The Ir-ReOx/SiO2 (4 wt% Ir, Re/Ir = 1, 2) showed high catalytic activity of C-O hydrogenolysis of glycerol, and it is characteristic that Ir-ReOx/SiO2 exhibited higher selectivity to 1, 3-propanediol than Rh-ReOx/SiO2 [32,33]. The structural change of Ir-ReOx/SiO2 (4 wt% Ir, Re/Ir = 1) during TPR with H2 was also investigated using in situ Ir and Re L3-edge quick-scanning X-ray absorption fine structure in the temperature range from room temperature to 900 K [37]. The valence of Ir and Re on Ir-ReOx/SiO2 (4 wt% Ir, Re/Ir = 1) was determined individually from the coordination number of Ir–O or Re-O bond in the EXAFS analysis and the chemical shift of the binding energy in the Ir and Re L3-edge XANES analysis [37]. It is concluded that Ir and Re species on Ir-ReOx/SiO2 (4 wt% Ir, Re/Ir = 1) are reduced almost simultaneously to metallic Ir and low valence Re (about +2) [37]. An interesting point is that the average valence of Re is maintained to be around 2+ in the temperature range of 485–600 K, and the further reduction of Re cation to metallic Re can be suppressed, although the reduction of Re is strongly promoted by the presence of Ir [37]. The coordination number of Re-Ir or Re bond increased clearly at higher reduction temperature (>600 K). The Re L3-edge EXAFS analysis indicates that the Re-O, Re-Ir or Re bonds are detected on Ir-ReOx/SiO2 (4 wt% Ir, Re/Ir = 1) [37]. In particular, the length of Re-Ir or Re bond is about 0.272–0.274 nm, which is also close to the sum of metal bond radius of Ir and Re, indicating the direct bond [37]. Based on the average valence of Re (~2+), coordination numbers and other characterization results, we propose that the Ir metal particles covered with 3-dimensional clusters of low valence Re oxides [37].
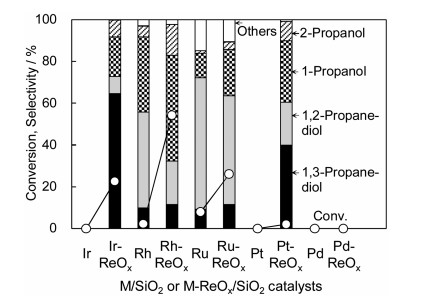
On the three catalysts of M-ReOx/SiO2 (M = Rh, Pt, Ir) catalysts, it is common that small clusters of low valent Re species is formed on the surface of metal particles, which can be explained by the higher oxygen affinity of Re than that of Rh, Pt and Ir. However, the catalytic performance in the C-O hydrogenolysis is strongly dependent on noble metals. The activity of Rh-ReOx/SiO2 and Ir-ReOx/SiO2 in the glycerol hydrogenolysis was much higher than that of Pt-ReOx/SiO2 (Fig. 6) [33].

|
Download:
|
| Fig. 6. Hydrogenolysis of glycerolover M/SiO2 and M-ReOx/SiO2(4 wt%M, Re/M = 0.25) catalysts. Conditions: glycerol 4 g, water 2 g, catalyst (reduced at 473 K (for Rh-ReOx/ SiO2: 393 K)) 0.15 g, H2SO4 H+/Re = 1, H2 8 MPa, 393 K, 12 h [33]. | |
{kind=link}
At present, this can be interpreted by the formation of active hydrogen species (such as hydride) on Rh-Re and Ir-Re interface from the heterolytic dissociation of H2. In contrast, high selectivity to 1, 3-propanediol in the glycerol hydrogenolysis can be related to the structure of Re oxide clusters, and 3-dimensional clusters are more favorable than 2-dimensional ones [8]. The rate-determining step can be the SN2-like attack of hydride at the interface of Rh-Re or Ir-Re to the adsorbed glycerol species on ReOx clusters as shown in Fig. 7. The presence of Re species at the second layer of the 3-dimensional clusters stabilize the adsorbed glycerol species giving 1, 3-propanediol. The mechanism of glycerol hydrogenolysis on Rh-ReOx and Ir-ReOx have been recently studied in a theoretical approach, and further investigation is necessary for the deeper understanding [49]. Rh-ReOx and Ir-ReOx catalysts are applicable to the selective and deep C-O hydrogenolysis of various substrates and selective hydrogenation of C = O in unsaturated aldehyde and so on (Fig. 8) [24-48,50,51].

|
Download:
|
| Fig. 7. Proposed mechanism of the hydrogenolysis of glycerol and related substrates over ReOx-modified Rh or Ir catalysts. | |
{kind=link}

|
Download:
|
| Fig. 8. Hydrogenation orhydrogenolysis catalyzedbyRh-ReOx/SiO2 andIr-ReOx/SiO2. | |
{kind=link}
The additive effect of Ni, Co, Zn, Cu, and Ag to Ir-ReOx/SiO2 (Re/Ir = 2) was investigated in the glycerol hydrogenolysis [41], and the addition of the above components decreased the catalytic activityofIr-ReOx/SiO2 drastically.The reasonof theactivitydecrease is not elucidated in detail, however, the above additives can be regarded as a poison for the hydrogen activation on Ir-ReOx/SiO2. For example, the above components have lower reducibility than noble metals, and they also tend to leach to the aqueous solution and the re-deposition on the catalyst surface. Judging from these results and behaviors, it is a challenging task to substitute noblemetals with non-noble metals in the C-O hydrogenolysis using the proposed reaction mechanism.
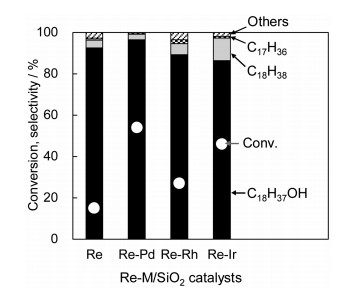
4. Re metal particle modified with low valent ReOx species for hydrogenation of carboxylic acidIn the X-ray absorption spectroscopic studies of Rh-ReOx/SiO2, the structural change of ReOx/SiO2 (3.6 wt% Re) catalyst during TPR with H2 was also investigated as a reference [31]. The reduction of ReOx/SiO2 proceeded above 600 K, which was much higher temperature than that in the presence of noble metals [31]. After the reduction at 873 K, the XRD peaks assigned to Re metal were detected clearly, and the particle size of Re metal was estimated to be 9.5 nm [31]. Assuming all the Re species are reduced to metallic state, the dispersion is calculated to be 0.14. At the same time, the coordination number of Re-Re bond is determined to be 10.9, which also supported the formation of rather large Re metal particles [31]. On the other hand, the dispersion from the amount of CO adsorption on ReOx/SiO2 was measured to be < 0.01, which is much lower than that from XRD, indicating that the CO adsorption is strongly suppressed [31]. One explanation for this disagreement in the dispersion is covering Re metal surface with low valent Re species. The presence of low valent Re species is supported by the non-zero average valence from XANES analysis (0~+1) and the presence of Re-O bond from the EXAFS analysis [31]. It is characteristic that high oxophilicity of Re element enables the coexistence of metallic and cationic states, which is connected to unique catalytic properties of Re. On the other hand, the coexistence of various states of Re species makes the structural elucidation difficult. We discussed and proposed the structure of Re-Pd/SiO2, where Re is the main catalytically active component and Pd is the cocatalyst, for the hydrogenation of carboxylic acid. This is supported by the results that Pd/SiO2 showed almost no activity, and monometallic Re/SiO2 showed some activity, and bimetallic Re-Pd/SiO2 showed clearly higher activity than Re/SiO2 at the same Re loading amount [52-54]. In addition, the combination of Pd with Re (Re-Pd) is more suitable to that of Re-Rh and Re-Ir from the viewpoint of the target product yield (alcohols), which can be related to lower reactivity of the target products (alcohols) on Pd-ReOx than that on Rh-ReOx and Ir-ReOx in the C-O hydrogenolysis (Fig. 9).

|
Download:
|
| Fig. 9. Hydrogenation of stearic acid overRe-M/SiO2 catalysts. Conditions: stearic acid 1 g, 1, 4-dioxane19 g, catalyst (14 wt%Re, M/Re = 0or1/8)0.1 g, H2 8 MPa, 413 K, 4 h [52]. | |
{kind=link}
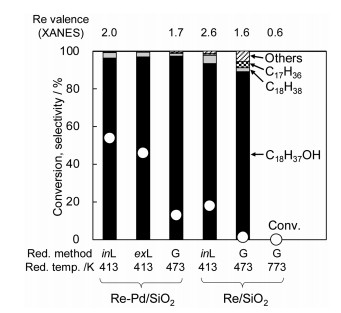
Another important factor for high catalytic performance in the hydrogenation of carboxylic acids over Re-Pd/SiO2 catalysts is the reduction method (Fig. 10) [53]. In our case, we compared the three reduction pretreatment methods: gas phase reduction, in-situ liquid phase reduction, ex-situ liquid phase reduction. Here, "in-situ" means liquid phase containing both substrate (carboxylic acid) and solvent (1, 4-dioxane), and "ex-situ" means liquid phase containing only the solvent, and in all cases, H2 is the reductant.

|
Download:
|
| Fig. 10. Effect of reduction method on hydrogenation of stearic acid over Re-Pd/SiO2 catalysts. Conditions: stearic acid 1 g, 1, 4-dioxane 19 g, catalyst (14 wt% Re, Pd/Re = 0 or 1/8) 0.1 g, H2 8 MPa, 413 K, 4 h. For reduction method, "inL", "exL" and "G" means in situ reduction in liquid phase, ex situ reduction in liquid phase, and gas phase reduction, respectively [53]. | |
{kind=link}
As shown in Fig. 10, gas phase reduction at 473 K is not suitable on both catalysts. According to the TPR profiles, the reduction of Re/SiO2 needs higher temperature than Re-Pd/SiO2, therefore, we also tested the gas phase reduction at 773 K, Re/SiO2 showed low catalytic activity. On the other hand, Re/SiO2 with in-situ liquid phase reduction showed rather high catalytic activity. Both Re-Pd/SiO2 with in-situ and ex-situ liquid phase reduction showed much higher activity than that with gas phase reduction and ReOx/SiO2 with in-situ liquid phase reduction [53].
In the XRD patterns of the catalysts after the catalytic use, which were measured without exposing the samples to air, the peaks assigned to Re hcp metal and Re fcc metal are detected, Based on the area of peaks and the dispersion estimated from the peak width, the number of Re metal atoms on the used catalysts can be estimated by the sum of (the area) x (the dispersion), which is called the effective XRD area. Fig. 11 shows the relation between this effective XRD area and conversion rate of the hydrogenation of stearic acid [53]. Regarding Re-Pd catalysts with the in-situ liquidphase reduction, the conversion rate can be related to effective XRD area, however, the conversion rate on Re-Pd and Re catalysts with gas phase reduction was clearly lower than that on Re-Pd catalysts with the in-situ liquid-phase reduction, suggesting that the surface structure or property of the catalysts with gas phase reduction is different from that of the catalysts with in-situ liquidphase reduction.

|
Download:
|
| Fig. 11. Conversion rate of stearic acid hydrogenation as a function of effective XRD area of Pd/SiO2 (1 wt% Pd), Re-Pd/SiO2 (1 wt% Pd) and Re/SiO2 (14 wt% Re) catalysts. (a) Pd (inL, 413, Reaction), (b) Re-Pd (inL, 413, Reaction) (Re/Pd = 2), (c) Re-Pd (inL, 413, Reaction) (Re/Pd = 4), (d) Re-Pd (inL, 413, Reaction) (Re/Pd = 6), (e) Re-Pd (inL, 413, Reaction) (Re/Pd = 8), (f) Re-Pd (inL, 413, Reaction) (Re/Pd = 12), (g) Re (inL, 413, Reaction), (h) Re-Pd (G, 473) (Re/Pd = 8), (i) Re (G, 473), (j) Re (G, 473). Effective XRD area = XRD area × Re dispersion. "Reaction" in the parenthesis represents the sample after the reaction of stearic acid hydrogenation. Reprinted with permission [53]. Copyright 2015, American Chemical Society. | |
{kind=link}
According to the Re L3-edge XANES analysis, the average valence of Re on the catalysts with gas phase reduction is relatively lower than that on the catalysts with in-situ liquid phase reduction (Fig. 10).The combinationof the characterization results enables the estimation of the ratio of (Re3++Re4+)/(Res0+Pds0) on Re-Pd/SiO2 (Re = 14 wt%, Pd = 1 wt%, Re/Pd = 8), where Ms0 represents the number of metal atom on the surface of the metal particles, and it is the degree of the coverage of Re cations on the metal surface. Highly active Re-Pd (exL, 413) in the hydrogenation of succinic acid and Re-Pd (inL, 413) in the hydrogenation of stearic acid gave 0.9 and 2.2 of (Re3++Re4+)/(Res0+Pds0), respectively, after the reaction. In contrast, high value (7.9) and very low value (0.4) of (Re3++Re4+)/(Res0+Pds0) over Re-Pd (inL, 413) and Re-Pd (G, 473) with lower catalytic activity were observed. From the comparison, the medium coverage of Re cations on the metal surface can be connected to high catalytic activity of the hydrogenation of carboxylic acids, which suggests that the interaction between Re cations and Re (or Pd) metal surface can be the catalytically active site.
5. ConclusionsHigh valent rhenium species stabilized on CeO2 modified with Pd and Au catalyzed the deoxydehydration of various substrates with vicinal OH groups with H2 reductant as heterogeneous catalysts. Medium valent rhenium species are stabilized on the surface of Rh and Ir metal particles, and the interface of Re cations and the metal surface can be a catalytically active site for the C-O hydrogenolysis of various biomass-related substrates. In addition, part of rhenium on SiO2 at high loading amount was reduced to metallic state to give Re metal particles and cationic Re species partially covered the surface of Re metal particles. Reduction conditions can influence the reduction degree of Re species and the coverage ratio of cationic Re species. The Re-Pd/SiO2 with medium coverage ratio of cationic Re species is suitable to the catalysts for hydrogenation of carboxylic acids. The control or stabilization of specific oxidation states of Re species will be connected to the development of catalysts with high performance.
| [1] |
Y. Nakagawa, K. Tomishige, Catal. Sci. Technol. 1 (2011) 179-190. DOI:10.1039/c0cy00054j |
| [2] |
Y. Nakagawa, K. Tomishige, Catal. Today 195 (2012) 136-143. |
| [3] |
Y. Nakagawa, M. Tamura, K. Tomishige, ACS Catal. 3 (2013) 2655-2668. |
| [4] |
Y. Nakagawa, M. Tamura, K. Tomishige, J. Mater. Chem. A 2 (2014) 6688-6702. DOI:10.1039/C3TA15384C |
| [5] |
K. Tomishige, M. Tamura, Y. Nakagawa, Chem. Rec. 14 (2014) 1041-1054. DOI:10.1002/tcr.201402026 |
| [6] |
Y. Nakagawa, S. Liu, M. Tamura, K. Tomishige, ChemSusChem 8 (2015) 1114-1132. DOI:10.1002/cssc.201403330 |
| [7] |
Y. Nakagawa, M. Tamura, K. Tomishige, Catal. Surv. Asia 19 (2015) 249-256. DOI:10.1007/s10563-015-9194-2 |
| [8] |
K. Tomishige, Y. Nakagawa, M. Tamura, Green Chem. 19 (2017) 2876-2924. |
| [9] |
Y. Nakagawa, M. Tamura, K. Tomishige, Res. Chem. Intermed. 44 (2018) 3879-3903. DOI:10.1007/s11164-018-3481-2 |
| [10] |
Y. Nakagawa, M. Tamura, K. Tomishige, Fuel Proc. Technol. 193 (2019) 404-422. |
| [11] |
J.J. Bozell, G.R. Petersen, Green Chem. 12 (2010) 539-554. DOI:10.1039/b922014c |
| [12] |
G.K. Cook, M.A. Andrews, J. Am. Chem. Soc. 118 (1996) 9448-9449. DOI:10.1021/ja9620604 |
| [13] |
M. Shiramizu, F.D. Toste, Angew. Chem. Int. Ed. 51 (2012) 8082-8086. DOI:10.1002/anie.201203877 |
| [14] |
A.L. Denning, H. Dang, Z. Liu, K.M. Nicholas, F.C. Jentoft, ChemCatChem 5 (2013) 3567-3570. DOI:10.1002/cctc.201300545 |
| [15] |
N. Ota, M. Tamura, Y. Nakagawa, K. Okumura, K. Tomishige, Angew. Chem. Int. Ed. 54 (2015) 1897-1900. DOI:10.1002/anie.201410352 |
| [16] |
N. Ota, M. Tamura, Y. Nakagawa, K. Okumura, K. Tomishige, ACS Catal. 6 (2016) 3213-3226. |
| [17] |
S. Tazawa, N. Ota, M. Tamura, et al., ACS Catal. 6 (2016) 6393-6397. DOI:10.1021/acscatal.6b01864 |
| [18] |
Y. Nakagawa, S. Tazawa, T. Wang, et al., ACS Catal. 8 (2018) 584-595. DOI:10.1021/acscatal.7b02879 |
| [19] |
Y. Xi, W. Yang, S.C. Ammal, et al., Catal. Sci. Technol. 8 (2018) 5750-5762. DOI:10.1039/C8CY01782D |
| [20] |
T. Wang, S. Liu, M. Tamura, et al., Green Chem. 20 (2018) 2547-2557. |
| [21] |
M. Tamura, N. Yuasa, J. Cao, Y. Nakagawa, K. Tomishige, Angew. Chem. Int. Ed. 57 (2018) 8058-8062. DOI:10.1002/anie.201803043 |
| [22] |
J. Cao, M. Tamura, Y. Nakagawa, K. Tomishige, ACS Catal. 9 (2019) 3725-3729. DOI:10.1021/acscatal.9b00589 |
| [23] |
T. Wang, M. Tamura, Y. Nakagawa, K. Tomishige, ChemSusChem (2019). DOI:10.1002/cssc.201900900 |
| [24] |
A. Shimao, S. Koso, N. Ueda, et al., Chem. Lett. 38 (2009) 540-541. DOI:10.1246/cl.2009.540 |
| [25] |
S. Koso, I. Furikado, A. Shimao, et al., Chem. Commun. (2009) 2035-2037. |
| [26] |
Y. Shinmi, S. Koso, T. Kubota, Y. Nakagawa, K. Tomishige, Appl. Catal. B 94 (2010) 318-326. DOI:10.1016/j.apcatb.2009.11.021 |
| [27] |
K. Chen, S. Koso, T. Kubota, Y. Nakagawa, K. Tomishige, ChemCatChem 2 (2010) 547-555. DOI:10.1002/cctc.201000018 |
| [28] |
Y. Amada, S. Koso, Y. Nakagawa, K. Tomishige, ChemSusChem 3 (2010) 728-736. DOI:10.1002/cssc.201000040 |
| [29] |
S. Koso, Y. Nakagawa, K. Tomishige, J. Catal. 280 (2011) 221-229. DOI:10.1016/j.jcat.2011.03.018 |
| [30] |
S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa, K. Tomishige, Appl. Catal. B 111- 112 (2012) 27-37. |
| [31] |
S. Koso, H. Watanabe, K. Okumura, Y. Nakagawa, K. Tomishige, J. Phys. Chem. C 116 (2012) 3079-3090. DOI:10.1021/jp2114225 |
| [32] |
Y. Nakagawa, Y. Shinmi, S. Koso, K. Tomishige, J. Catal. 272 (2010) 191-194. DOI:10.1016/j.jcat.2010.04.009 |
| [33] |
Y. Amada, Y. Shinmi, S. Koso, et al., Appl. Catal. B 105 (2011) 117-127. DOI:10.1016/j.apcatb.2011.04.001 |
| [34] |
Y. Nakagawa, X. Ning, Y. Amada, K. Tomishige, Appl. Catal. A 433-434 (2012) 128-134. DOI:10.1016/j.apcata.2012.05.009 |
| [35] |
K. Chen, K. Mori, H. Watanabe, Y. Nakagawa, K. Tomishige, J. Catal. 243 (2012) 171-183. |
| [36] |
Y. Amada, H. Watanabe, Y. Hirai, et al., ChemSusChem 5 (2012) 1991-1999. DOI:10.1002/cssc.201200121 |
| [37] |
Y. Amada, H. Watanabe, M. Tamura, et al., J. Phys. Chem. C 116 (2012) 23503-23514. DOI:10.1021/jp308527f |
| [38] |
K. Chen, M. Tamura, Z. Yuan, Y. Nakagawa, K. Tomishige, ChemSusChem 6 (2013) 613-621. DOI:10.1002/cssc.201200940 |
| [39] |
Y. Nakagawa, K. Mori, K. Chen, et al., Appl. Catal. A 468 (2013) 418-425. DOI:10.1016/j.apcata.2013.09.021 |
| [40] |
S. Liu, Y. Amada, M. Tamura, Y. Nakagawa, K. Tomishige, Green Chem. 16 (2014) 617-626. DOI:10.1039/C3GC41335G |
| [41] |
M. Tamura, Y. Amada, S. Liu, et al., J. Mol. Catal. A 388-389 (2014) 177-187. DOI:10.1016/j.molcata.2013.09.015 |
| [42] |
S. Liu, Y. Amada, M. Tamura, Y. Nakagawa, K. Tomishige, Catal. Sci. Technol. 4 (2014) 2535-2549. DOI:10.1039/C4CY00161C |
| [43] |
S. Liu, M. Tamura, Y. Nakagawa, K. Tomishige, ACS Sustain. Chem. Eng. 2 (2014) 1819-1827. DOI:10.1021/sc5001463 |
| [44] |
S. Liu, Y. Okuyama, M. Tamura, et al., ChemSusChem 8 (2015) 628-635. DOI:10.1002/cssc.201403010 |
| [45] |
S. Liu, Y. Okuyama, M. Tamura, et al., Green Chem. 18 (2016) 165-175. DOI:10.1039/C5GC02183A |
| [46] |
S. Liu, M. Tamura, Z. Shen, et al., Catal. Today 303 (2018) 106-116. DOI:10.1016/j.cattod.2017.07.025 |
| [47] |
L. Liu, S. Kawakami, Y. Nakagawa, M. Tamura, K. Tomishige, Appl. Catal. B 256 (2019) 117775. DOI:10.1016/j.apcatb.2019.117775 |
| [48] |
T. Ebashi, Y. Ishida, Y. Nakagawa, et al., J. Phys. Chem. C 114 (2010) 6518-6526. DOI:10.1021/jp911908c |
| [49] |
J.J. Varghese, L. Cao, C. Robertson, et al., ACS Catal. 9 (2019) 485-503. DOI:10.1021/acscatal.8b03079 |
| [50] |
M. Tamura, K. Tokonami, Y. Nakagawa, K. Tomishige, Chem. Commun. 49 (2013) 7034-7036. DOI:10.1039/c3cc41526k |
| [51] |
M. Tamura, K. Tokonami, Y. Nakagawa, K. Tomishige, ACSCatal. 6 (2016) 3600-3609. |
| [52] |
Y. Takeda, Y. Nakagawa, K. Tomishige, Catal. Sci. Technol. 2 (2012) 2221-2223. |
| [53] |
Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura, K. Tomishige, ACS Catal. 5 (2015) 7034-7047. DOI:10.1021/acscatal.5b01054 |
| [54] |
Y. Takeda, M. Tamura, Y. Nakagawa, K. Okumura, K. Tomishige, Catal. Sci. Technol. 6 (2016) 5668-5683. DOI:10.1039/C6CY00335D |