 2020, Vol. 31
2020, Vol. 31 


b Key Laboratory of Smart Drug Delivery(Ministry of Education), State Key Laboratory of Medical Neurobiology, Department of Pharmaceutics, School of Pharmacy, Fudan University, Shanghai 201203, China;
c University of Chinese Academy of Sciences, Beijing 100049, China
Drug delivery systems (DDSs) [1] are used to precisely deliver the drugs at the intended targets to improve the selectivity and efficiency. Except that, DDSs also play a crucial role in prolonging the blood circulation by optimizing the pharmaceutical profile [2]. Due to the development of the pharmaceutical industry, drugs can be classified into different categories, including small molecular drugs, biomacromolecules (such as proteins, peptides, nucleic acids) [3,4]. Up to date, various DDSs for cancer therapy have been designed to efficiently deliver and release the drugs to the targeted tissues or cells. These targeting systems mostly exploit the enhanced permeability and retention (EPR) effect of the tumors [5]. Except that, the overexpressing receptors on the surface of cancer cell membrane can also be targeted by designing ligand-functionalized DDSs [6,7]. However, the precise and timely drug release in the cancer cells or tissues is one of the major barriers for DDSs [8]. Premature drug release usually results in severe adverse effects while insufficient drug release at the targeted sites. The premature drug release can be reduced by increasing the stability of DDSs but over-stable DDSs induced deficient drug accumulation also affects the therapeutic efficiency. The development of the smart DDSs capable of efficiently and precisely releasing the drug at the targeted regions is, therefore, a great challenge. Based on their structures, various DDSs have been designed by conjugating drugs with antibodies (antibody-drug conjugates, ADCs) [9,10], small organic ligands (small molecule-drug conjugates, SMDCs) [11], polymers [12], or non-covalently encapsulating by nano-particles (NPs) [13], and hybrid nanogels [14]. In ADCs, SMDCs, polymer-drug conjugates and prodrug-based DDSs, which are formed by covalent bonds, cleavable linkers play a crucial role for facilitating drug release at the targeted sites [15,16].
Dynamic covalent chemistry based on the dynamic features of reversible covalent bonds [17], has been extensively applied in a variety of areas, such as medicine [18], material science [19], and chemical biology [20,21]. Dynamic covalent bonds formed under thermodynamic equilibrium are reversible, tunable, and controllable [17]. Generally, the equilibrium can be disturbed by physical/ chemical stimuli (such as temperature, irradiation, and pH), which indicate the stimuli-responsive properties of dynamic covalent bonds. These features can be exploited for designing stimuliactivatable DDSs [22]. Dynamic covalent bond-based materials with stimuli-responsive linkers or functional groups (i.e., boronic acid) can be used as the carriers [23,24] or specific targeting ligands [25]. Therefore, dynamic covalent chemistry ensures the development of adaptive, flexible, and stimuli-responsive DDSs, which remain stable under physiological conditions. By improving the drug release kinetics, the dynamic covalent chemistry-based DDSs also can show improved efficiency for cancer therapy.
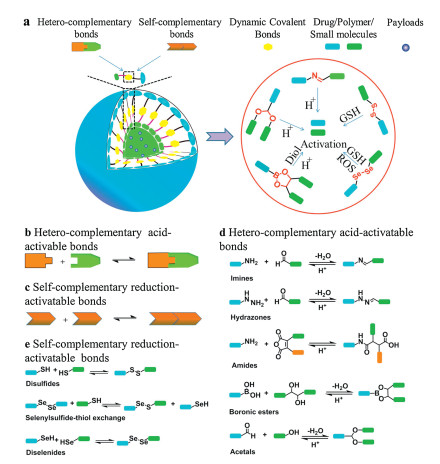
The dynamic covalent bonds can be generally divided into five main categories based on the composition of dynamic covalent bonds, including C–N, C–O, B–O, S–S, and Se-Se bonds (Fig. 1). Other frequently used bonds in the establishment of DDSs include C–Se bonds [26], C–S bonds [27], as well as C–C bonds [28], which are photoirradiation- or thermo-responsive as they are formed by the Diels-Alder reaction under irradiation or heat [29,30]. Dynamic covalent bonds can also be classified based on their responses to the particular stimuli, such as pH-responsive, reduction-responsive, and irradiation-responsive bonds [31,32]. In this review article, we will summarize the emerging applications of five categories of stimuli-responsive dynamic covalent bonds in the development of DDSs for cancer therapy.

|
Download:
|
| Fig. 1. Main classes of dynamic covalent bonds used in the design of DDSs. (a) Illustration of stimuli-responsive drug delivery systems; (b) Schematic representation of hetero-complementary acid-activatable bonds; (c) Schematic representation of self-complementary reduction-activatable bonds; (d) Representative types of hetero-complementary acid-activatable dynamic covalent bonds; (e) Representative types of self-complementary reduction-activatable dynamic covalent bonds. | |
2. Drug delivery systems based on acid-activatable dynamic covalent bonds
Dynamic covalent bonds can be cleaved under various stimuli, including acidic conditions. The well-established acidic-responsive dynamic covalent bonds are imines (C–N), acetals (C–O), and boronic ester (B–O). As the comparatively higher lactic acid concentration and hypoxic conditions in the tumor induce the acidic microenvironment [33], the acid-activatable dynamic covalent bond-based DDSs exploit the inherited tumor acidic microenvironment for accurate drug release, which might improve the efficiency of cancer treatment.
2.1. C–N bondsC-N bonds are the most common type in the dynamic covalent bonds and can be classified as imines, hydrazones, amides, and oximes, etc., based on their functional groups. All these linkages are formed by condensing the respective aldehydes with amines, including hydroxylamine, hydrazine, etc., and hydrolyzed under acidic environment. Therefore, the C–N dynamic covalent bonds are extensively applied in the formulation of acid-activatable DDSs [34].
2.1.1. Imines (Schiff bases)Imines, also known as Schiff bases [35-38], are widely used in chemical synthesis. Imines are one of the most classical and acid-responsive dynamic covalent bonds, which have been applied as linkers in acid-responsive DDSs. For example, Nie et al. designed magnetic nanoclusters (NCs), which were modified with the antibody against programming death-1 (anti-PD-1 antibody) through the benzoxic-imine bonds [39]. As the anti-PD-1 antibody could bind to the effector T cells specifically, therefore the T cells could be delivered to the tumor site with the antibody-modified NCs under the guidance of magnetic resonance imaging (MRI). In the weak acidic tumor microenvironment, the imine bonds were hydrolyzed and released the ant-PD-1 antibody, which worked synergistically with the effector T cells for improving the performance of adoptive T-cell therapy by enhancing the intratumoral accumulation of the effector T cells.
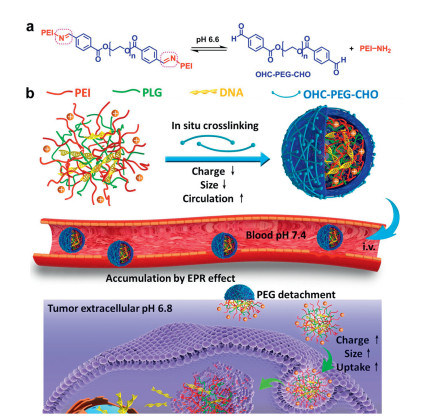
Generally, the imine-containing materials are formed through the condensation reactions between amino-functional polymer and aldehyde, and these acid-responsive materials can be used to encapsulate the payloads in the core of the self-assembled NPs. For instance, Guan et al. designed an ultrasensitive pH-responsive gene delivery system, namely P[(GP)D], which was composed of poly-L- glutamate (PLG) and polyethylenimine (PEI) and then crosslinked with aldehyde-modified PEG through the imines (Fig. 2) [40]. And the DNA was loaded in the core of the P[(GP)D] polyplexes through electrostatic interactions, while the polymer cross-linking shielded the polyplexes. The cross-linked polyethylene glycol (PEG) not only improved the stability by shielding the positive charge, but also extended the circulation time of the NPs. When NPs arrived in the tumors (acidic tumor microenvironment), the PEG corona was peeled off rapidly via hydrolysis of the imine bonds, which enhanced the tumor uptake efficiency by expanding size/charge of NPs. The prepared polyplexes displayed ultra-sensitivity to the acidic environment as the particle size and cellular uptake dramatically changed at pH 6.8. Then the VEGF-targeted plasmid DNA can be accumulated in the tumor cells to inhibit angiogenesis in tumor. Therefore, this gene delivery system improved the antitumor efficiency in vivo.

|
Download:
|
| Fig. 2. Schematic illustration of the imine-based P[(GP)D] NPs. (a) The acid-responsive mechanism of the Schiff base in P[(GP)D] NPs; (b) The mechanism of the imine-based P[(GP)D] gene delivery system. Copied with permission [40]. Copyright 2016, American Chemical Society. | |
Similarly, Han et al. constructed a PEGylated polyethyleneimine-coated gold NP-based tumor microenvironment-responsive nanosystem to co-deliver all-trans retinoic acid (ATRA) and heat shock protein 47 (HSP 47)-targeting siRNA [41]. With the reeducation of pancreatic stellate cells (PSCs) and inhibition of extracellular matrix hyperplasia by the tumor-targeted NPs, the microenvironment of pancreatic can be rebuild, which help to improve the efficiency of chemotherapeutics in pancreatic cancer. Hu et al. also designed imine-containing aminoglycoside hydrogels by exploiting the interaction between the aminoglycoside and aldehyde-functionalized dextran [42,43].
2.1.2. HydrazonesHydrazone linkages are relatively more complex than the imines in structure as they are formed by the condensation of aldehyde and hydrazine. Hydrazone is also one of the classical C–N dynamic covalent bonds which are widely applied in the fabrication of DDSs [44]. Generally, hydrazones are introduced into the drug conjugates as linkers to provide the acid-activatable property [45]. Among the ADCs on the market, both Mylotarg (gemtuzumab ozogamicin) [46] and Besponsa (inotuzumab ozogamicin) [47] have introduced the hydrazone into the molecule to conjugate the antibody with ozogamicin. When the ADCs are uptaked by the tumor cells, the conjugated ozogamicin can be released in the intracellular acidic microenvironment for tumor regression.
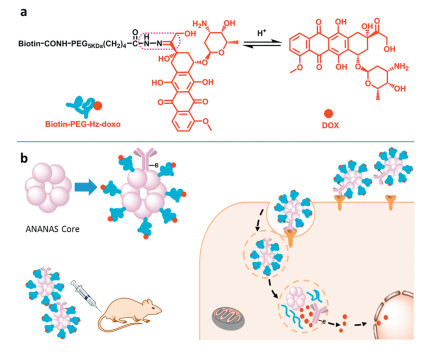
Except introducing the hydrazone linkage through other molecules, the hydrazone bonds also can be formed by the aldehyde/ketone or hydrazine groups that belong to the drug, for instance, doxorubicin (DOX), which has ketone groups in the molecular structure, can be used in this way. For instance, Sedlacek et al. demonstrated a drug delivery system, where the DOX was conjugated to the polymer backbone via hydrazone linkage [48], which triggered drug release in the acidic tumor microenvironment and thus enhanced cellular uptake and antitumor efficiency. Similarly, Pearce et al. designed a prostatespecific membrane antigen (PSMA)-targeted hyperbranched polymer-DOX conjugates through hydrazone linkage [49]. Furthermore, Roncato et al. designed an anti-EGFR antibody (cetuximab)-functionalized NPs, which used the avidin-nucleicacid-nano-assemblies (ANANAS) as the core that can be decorated by the biotinylated antibody and worked as the carriers of hydrazone-containing DOX-biotin conjugates (Fig. 3) [50]. As the ANANAS Nps were decorated by the anti-EFGR antibody, the cellular uptake of NPs was promoted in the MD-MBA-231 (Triple Negative Breast Cancer, TNBC) by the EGFR-mediated internalization. DOX can be released from the NPs efficiently since the hydrazone linkage between DOX and pegylated biotin can be cleaved in the acid environment of lysosomes, which improved the anti-tumor efficiency of DOX.

|
Download:
|
| Fig. 3. Schematic illustration of the imine-based anti-EFGR antibody-guided avidin-nucleic-acid-nano-assemblies (ANANAS). (a) The acid-responsive mechanism of the Biotin-PEG-DOX conjugates; (b) The drug release mechanism of the hydrazone-based antibody-modified ANANAS. Reproduced with permission [50]. Copyright 2018, Springer Nature. | |
2.1.3. Amides
Amides are of paramount importance in the C–N covalent bonds. These bonds are mostly stable and can only be hydrolyzed in harsh conditions. However, some amide linkers (such as cis-aconitamide, citraconamide, and maleamide) comprised of derivatives of maleic acid can be acid-responsive, which made them promising dynamic covalent bonds that can be applied in the acidactivatable DDSs. For example, Li et al. reported pH-responsive clustered NPs comprised of acid-labile amide linkage [51]. As the clustered NPs were formed by polycaprolactone-modified cisplatin prodrug-conjugated poly(amidoamine) dendrimers through the connection of 2-propionic-3-methylmaleic anhydride (CDM), the cisplatin prodrug-dendrimers were therefore released in the acidic tumor microenvironment and penetrated into the deep tumor tissue efficiently. With the enhanced uptake by the tumor cells, the cisplatin can be restored from the prodrug by glutathione (GSH) reduction to perform antineoplastic activity.
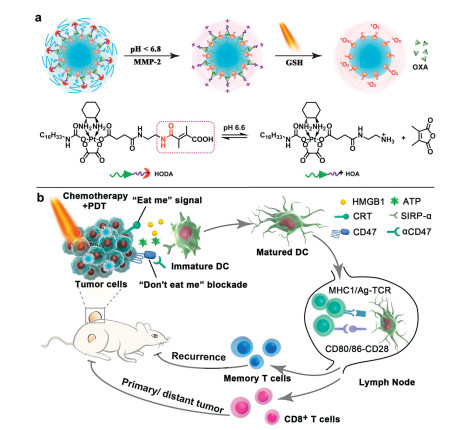
Zhou et al. designed a delivery system (MPV-HOAD) to codeliver anticancer drug oxaliplatin (OXA) and a photosensitizer pheophorbide a (PPa) by combining the maleic acid-derived OXA prodrug and an enzyme-responsive derivative of PPa. Thus, MPV-HOAD can be activated by the enzymatic and acidic tumor microenvironment (Fig. 4) [52]. 2, 3-Dimethylmaleic anhydride (DMMA) was triggered to release OXA prodrug in the acidic microenvironment. Meanwhile, the PEG shielding layer can be peeled off with endogenous metalloproteinase-2 (MMP-2), which can enhance the tumor penetration and cellular uptake with the reversal of the surface charge of NPs synergistically. OXA can be released by the reduction with GSH and work together with PPabased photodynamic therapy to induce the immunogenic cell death (ICD) and reprogram the immunosuppressive tumor microenvironment. Combined with the anti-CD47 antibody (αCD47), MPV-HOAD can inhibit the primary tumor growth and suppress distant metastasis of tumor cells efficiently [53,54].

|
Download:
|
| Fig. 4. Schematic illustration of the acid-activatable MPV-HOAD NPs. (a) The acid-responsive mechanism of the maleic amide-containing amide bond-based MPV-HOAD NPs; (b) The immunoregulation mechanism of MPV-HOAD NPs. Copied with permission [52]. Copyright 2019, Wiley Publishing Group. | |
2.1.4. Oximes
Similar to the imines and hydrazones, oximes can also be used to construct acid-activatable DDSs [55]. For example, O'Brien et al. designed a method to deliver the nucleic acids into cells efficiently through the cell surface engineering with nucleic acid/lipid nanocomplex by oxime bonds, which might be used for gene therapy of cancer (Fig. 5) [56]. With the liposome fusion strategy, the tailoring cell surfaces were engineered by ketone, which can be interacted with the nucleic acid complex bio-orthogonal liposomes rapidly. Then the lipoplex can be endocytosed and released the nucleic acids efficiently in the intracellular acid microenvironment of the primed cells. Thus, the nucleic acids can be efficiently delivered into the primed cells, which provided the new options for improved cancer treatment.

|
Download:
|
| Fig. 5. Schematic illustration of the cell surface engineering strategy for the gene delivery. (a) The acid-responsive mechanism of the oxime linkage in the cell engineering strategy; (b) The mechanism of the cell surface engineering strategy. Copied with permission [56]. Copyright 2017, American Chemical Society. | |
2.2. C–O bonds
Compared to C–N bonds, C–O bonding linkages are relatively less utilized in the dynamic covalent chemistry due to their stabilities. C–O bond-based dynamic covalent bonds can be classified as hemiacetals, acetals, orthoesters, and esters, etc. Among all, acetals, and orthoesters are acid-responsive dynamic covalent bonds that can be formed by reversible nucleophilic reaction with C = O bonds under acidic conditions, while esters are so stable that only can be hydrolyzed under strong acidic/basic condition, and hemiacetals are the intermediates of acetals as they can convert to acetals under thermodynamic equilibrium spontaneously. Thus, the acetals and orthoesters are generally used in the designing of C–O dynamic covalent bondbased DDSs.
Acetals/ketals are formed by the nucleophilic reactions of alcohols with aldehydes or ketones, and hydrolyzed in mild acid conditions, thus can be used as pH-responsive linkers for the fabrication of stimuli-responsive DDSs [57,58]. For example, Zhong et al. demonstrated acetals as linkers for the synthesis of paclitaxel (PTX) prodrug micellar NPs [59,60], in which the hydroxyl group of PTX was connected with dendritic oligo glycerol-functionalized hyaluronic acid or PEG-block-poly(acrylic acid) through the acetal bonds. PTX was released from the prodrug micellar NPs through the cleavage of acetals under the endosomal acidic conditions and exhibited the antitumor efficiency. In a similar study, Guo et al. designed the estradiol-polyketal conjugates to deliver estradiol [61].
Dextran contains natural acetal groups and multiple hydroxyl groups which can be used to formulate the acetals, therefore, can be used as pH-responsive carriers in acidactivatable DDSs. For example, Bachelder et al. presented acetal-modified dextran as pH-responsive DDSs [62]. The modifications of acetals enabled the amphiphilic acetal-dextran to self-assemble into NPs and then hydrolyzed into the watersoluble dextran in the acidic microenvironment. Due to its pH-responsive properties, the acetal-dextran has been applied for the delivery of nucleic acids [63,64] and proteins [65]. The cellular uptake also can be enhanced by acetal-dextran-modified NPs for highly efficiently delivery of payloads. Zou et al. developed a nanomedicine DDSs, so called Ang-RBCm@NM- (Dox/Lex), by combing pH-sensitive property of acetal-dextran with red blood cell membranes, which was further functionalized with the angiopep-2 for co-delivery of DOX and lexiscan (Lex) [66]. NPs based on acetal-dextran prolonged the blood circulation, enhanced blood-brain-barrier penetration, improved accumulation in tumor, and achieved pH-sensitive drug (Dox and Lex) release thus improved the antitumor efficiency.
In addition to the synthetic acetal-modified dextran, the oxidation or reduction products of dextran, i.e., poly(1- hydroxy-methylethylene hydroxymethyl-formal) (PHF) [67], can also be used for the preparation of pH-responsive DDSs owing to the presence of intrinsic acetal groups in the dextran derivatives. Based on the intrinsic acid-responsive property of PHF, Mersana et al. designed a series of pH-responsive PHFbased DDSs of ADCs [68]. Kumazawa et al. also reported an acidliable polymeric prodrug by conjugating exatecan with carboxymethyl dextran polyalcohol (CM-Dex-PA, modified dextran) [69]. Owing to the presence of pH-responsive dextran or dextran derivatives, the DDSs can acquire long-term retention and stimuli-responsive drug release in tumor microenvironment and ultimately enhance the antitumor efficiency. Except dextran, other polysaccharides, like β-cyclodextrin [70] can also be modified in a similar manner.
In addition to acid-liable acetal bonds, orthoester bonds have been exploited for engineering stimuli-activatable DDSs [71,72]. For instance, Yan et al. designed a series of poly(orthoester)-based acid-responsive drug carriers to deliver chemotherapeutics and showed satisfying antitumor efficiency in xenograft tumor-bearing mouse model [73].
2.3. B–O bondsBoronic acid and boronic ester play key roles in the chemical synthesis as they are the reactants of Suzuki coupling, which are used to form new C–C bond linkages. Moreover, boronic acids are extensively used in the molecular recognition and DDSs [74,75] owing to their ability to undergo reversible reactions with the Lewis base (like diols). Generally, five or six-membered ring structure containing boronic ester are formulated through the interaction between boronic acid and 1, 2-diols or 1, 3-diols respectively. As the boronic esters can be hydrolyzed or reformed under diols and mild acid environment, they are dual-responsive (acid and diol) dynamic covalent bonds. Therefore, these linkages are exploited to design acid-activatable and diols-responsive DDSs [76-81].
The boronic esters are generally formed between the boronic acids with the 1, 2-diols or 1, 3-diols structure in saccharides. Based on this strategy, a wide range of DDSs have been reported [82], such as the gel-based insulin delivery system which can regulate the concentration of blood glucose through the reformulation of boronic ester [83]. Sialic acid as a natural carbohydrate which is overexpressed on the surface of cancer cells and can interact with boronic acid-containing compounds or polymers, can be the target for the tumor-targeting drug delivery systems to improve the cellular uptake of the therapeutics. A variety of boronic acidcontaining polymers are therefore designed as drug carriers for the tumor targeting therapy.
In addition to saccharides, boronic acid can also interact with phenols like catechol [84,85]. Boronic acid-containing molecules, diols are used to fabricate boronic ester-based DDSs. For example, Liu et al. designed a kind of polydopamine NPs which was PEGylated and borate-coordination polymer-coated [86]. PEG was sheddable in the mild acid tumor microenvironment due to the presence of boronic ester conjugation in dopamine-modified PEG containing NPs. Moreover, the interaction between free boronic acid groups and overexpressed sialic acid further enhanced the cellular uptake and antitumor efficiency.
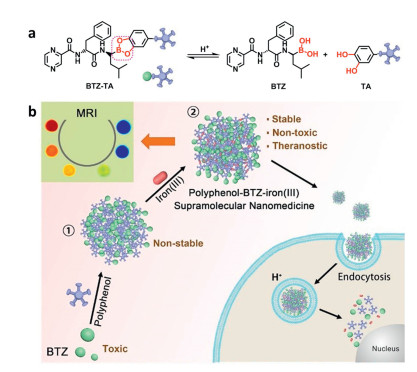
Wang et al. designed a new class of supramolecular nanomedicine by using Bortezomib (BTZ, a boronate proteasome inhibitor) as the loading drug (Fig. 6) [87]. In this delivery system, BTZ was conjugated with the catechol-containing natural polyphenols via the boronate ester bond. The ferric ions were incorporated with polyphenol to further stabilize the NPs as well as to introduce the bioimaging functionality. NPs were degraded and released the BTZ in the acidic microenvironment of tumor cells after being endocytosed. The proposed delivery system thus exhibited the antitumor profile without inducing systematic sideeffects.

|
Download:
|
| Fig. 6. Schematic illustration of the formation of polyphenol-BTZ-iron(Ⅲ) supramolecular nanomedicine. (a) The acid-responsive mechanism of the boronic ester bond of the polyphenol-BTZ-iron (Ⅲ) supramolecular nanomedicine; (b) The composition and drug release mechanism of the polyphenol-BTZ-iron(Ⅲ) supramolecular nanomedicine. Copied with permission [87]. Copyright 2018, American Chemical Society. | |
3. Drug delivery systems based on reduction-activatable dynamic covalent bonds
Except the mild acid microenvironment, abundant GSH in the tumor cell also creates a reduction environment [88] which can be exploited as stimuli to cleave disulfide and diselenide bonds in tumor. Therefore, reduction-responsive DDSs based on the disulfides and diselenide linkages have been established.
3.1. S–S bondsDisulfides, the well-studied dynamic covalent bonds, which can stabilize protein structure during protein folding, are ubiquitous in living bodies. In contrast to imines and acetals, disulfides bonds are reduction-responsive. Due to their GSH-responsive properties, disulfides linkages are frequently used for the design of tumortargeted DDSs [89-93].
Similar to the other dynamic covalent bonds, disulfides can be used as cleavable linkers in the drug conjugates or to formulate stimuli-responsive carriers. Various disulfides linker-based ADCs are in clinical trials [94]. Due to GSH reduction, the payloads can be released in the tumor cells. Qi et al. reported a polymer-drug conjugate of monomethyl auristatin E (MMAE), which used disulfide as the linker of polymer and MMAE [95]. The mPEG-b-PGA-coated self-assembled NPs of polymer-drug conjugates were stable in the normal physiological environment. Upon cellular uptake, mPEG-b-PGA was dissociated from the core of NPs in the acid microenvironment of endosomes/lysosomes, and thus released MMAE. The conjugates displayed improved the antitumor efficiency in a patient-derived xenograft (PDX) tumor model of advanced ovarian cancer.
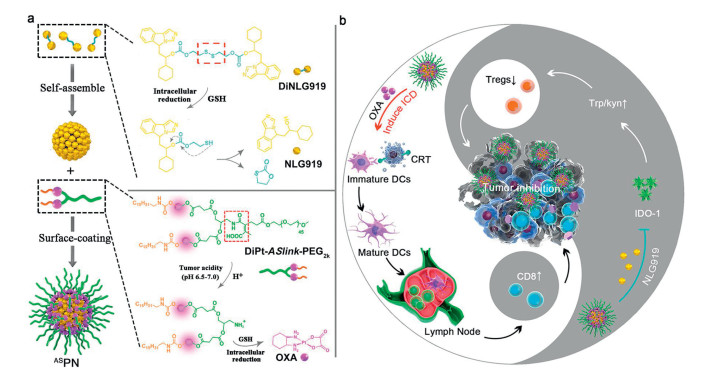
Feng et al. reported a light-inducible nanocargo (LINC) which was formulated by OXA prodrug and a disulfide-containing heterodimer that composed of PPa and NLG919 (a potent IDO-1 inhibitor) for combination immunotherapy (Fig. 7) [96]. As the NPs can accumulate in the tumor via the EPR effect, the first irradiation in the tumor site would induce the reactive oxygen species (ROS) generation to trigger the cleavage of the PEG corona, which can promote the penetration of LINC in tumor. With the interaction of GSH in the tumor cells, OXA, PPa and NLG919 were restored and worked together to reverse the immunosuppressive tumor microenvironment and improve the antitumor efficiency.

|
Download:
|
| Fig. 7. Schematic illustration of the light-inducible nanocargo LINC. (a) The reduction-responsive mechanism of the disulfides bonds containing heterodimer; (b) The immunoregulation mechanism of LINC. Copied with permission [96]. Copyright 2019, Wiley Publishing Group. | |
The desired cargo also can be conjugated with the biodegradable material by disulfide linkages. Yuan et al. designed a delivery system by conjugating native antibodies with biodegradable silica nanoquencher, and further coated with a cell-penetrating poly(disulfides) [97]. Therefore, the cellular uptake was enhanced via thiol-mediated pathways and ultimately triggered the release of the antibody in the tumor microenvironment while escaping the endolysosomal barriers. Ling et al. also designed a Pt(Ⅳ) delivery system comprising of poly(disulfide amide) polymer and lipidmodified PEG [98]. In this system, the poly(disulfide amide) polymer scavenged the GSH in the tumor cells and overcame the cisplatin resistance.
3.2. Se–Se bondsSelenium shares some characteristics with sulfur since both belong to the same group of the periodic table. However, selenium is relatively less electronegative due to the larger atomic radius, which makes diselenide a promising dynamic covalent bond with lower Se–Se bond energy.
Selenium is an amphoteric element. In a reversible process, diselenides can be reduced to selenols by GSH while oxidized to selenic acid by the H2O2 or ROS in an irreversible reaction [99]. Owing to stimuli-responsive, such as GSH and ROS-responsive features in the tumor microenvironment, diselenides have a great potential in DDSs either as reduction-responsive or the oxidation-responsive systems. For example, Sun et al. designed a drug delivery system formulated by the diselenide bond-containing paclitaxel-citronellol (PTX-CIT) conjugates and DSPE-PEG (Fig. 8) [100]. When the NPs were uptaken by the tumor cells, the diselenides can be broken by the highly concentrated GSH or ROS microenvironment in the tumor cells, which can promote the release of PTX in the cells, and eventually improve the antitumor efficiency of the drugs. Compared to other sulfur/selenium/carbon linkage-based prodrugs, diselenide-containing DDSs showed the most potent antitumor efficiency as the diselenide bonds can be cleaved by the oxidation or reduction environment in the tumor sites. Li et al. reported a co-delivery system of let-7b antisense oligonucleotide, superparamagnetic iron oxide nanocubes (SPIONs), and Simvastatin, in which the diselenide bond was used as a linker to design a poly-prodrug by conjugating polymers with Simvastatin [101].

|
Download:
|
| Fig. 8. Schematic illustration of the formulation of diselenide-bridged PTX-CIT conjugate nanoassemblies. (a) The dual responsive mechanisms of the diselenidebridged conjugate nanoassemblies; (b) The PTX release mechanism of the nanoassemblies. Copied with permission [100]. Copyright 2019, Springer Nature. | |
Shao et al. also fabricated a biodegradable mesoporous silica NPs (MSNs) by introducing a diselenide-containing organosilica moiety [102]. Cytotoxic RNase A was encapsulated in diselenidebridged MSNs to precisely deliver the payload (RNase A) under redox or oxidative conditions while MSNs exhibited homologous targeting and immune-invasion features due to cancer-cellderived membrane coating.
4. Drug delivery systems based on dual dynamic covalent bondsTo achieve accurate drug release at the tumor sites and improved cancer therapy, dual dynamic covalent bonds-based DDSs were also explored in last decades. Dual dynamic covalent bond linkages are capable of undergoing cleavages under various inherent stimuli of the tumor microenvironment and therefore can enhance the specificity of DDSs and avoid adverse side-effects.
Feng et al. demonstrated a dual-responsive binary cooperative prodrug NP (BCPN) drug delivery system, which is comprised of PEG-modified OXA prodrug and NLG919 dimer (Fig. 9) [103], where PEG was conjugated with OXA by acid-activatable amide while core NPs were formed by the NLG919 dimer with the disulfide linker. In the tumor microenvironment, the IDO inhibitor NLG919 was released via reduction of the disulfide bonds, where OXA and NLG919 synergistically function to improve antitumor efficiency by regulating the immunosuppressive tumor microenvironment.

|
Download:
|
| Fig. 9. Schematic illustration of the BCPN NPs based on dual dynamic covalent bonds. (a) The formulation of BCPN NPs and the response mechanism of the prodrugs; (b) The immunoregulation mechanism of BCPN. Copied with permission [103]. Copyright 2018, Wiley Publishing Group. | |
Xue et al. also designed a dual transformability nanotheranostics through the application of hydrazone and imine linkages (Fig. 10) [104]. As both amine and ketone group are present in DOX, PPa-Hyd-DOX (PhD) dimer can be primely formed by hydrazidemodified PPa and DOX, and then the PhD dimer can further crosslink with aldehyde functionalized PEG by imine bonds to stabilize the NPs. When the NPs were injected into the mice, they can accumulate in the tumor through the EPR effect, then the NPs can be depegylated in the mild acid tumor microenvironment for the acid-activatable imine linkage, the PhD monomer also can permeate in the deep tumor tissue, not only enhanced the cell uptake, but can also be applied for the tumor imaging. After the endocytosis of tumor cells, the drugs can be released and exhibit the antitumor efficiency. Except that, DDSs can also be designed by the combination of disulfide and diselenide linkages based on their different sensitivity to the tumor microenvironment (such as oxidative and reductive-oxidative microenvironment) [105].

|
Download:
|
| Fig. 10. Schematic illustration of the dual transformability nanotheranostics. (a) The dual acid-responsive mechanism of the nanotheranostics; (b) The composition and drug release mechanism of nanotheragnostics. Copied with permission [104]. Copyright 2018, Springer Nature. | |
5. Conclusions and perspectives
The development of dynamic covalent chemistry has resulted in the development of various dynamic covalent linkages with the remarkable features like adaptability, specificity, flexibility, and stimuli-responsive capabilities. Up to date, lots of dynamic covalent bond-based systems have been extensively exploited for controlled drug delivery, in particular for cancer treatment (Table 1). Compared with the convenient DDSs with stable chemical structures, dynamic covalent bond-based DDSs display several distinct advantages. First, the dynamic covalent bonds can be prepared in a reversible manner under specific condition, which allows stable drug encapsulation while controllable drug release of the DDSs. Second, the formulation of the DDSs can be self-stabilized via the formation of thermodynamic equilibrium of dynamic covalent bonds. Furthermore, as most dynamic covalent bonds can be formed in mild conditions, the dynamic covalent bond-based DDS can be readily engineered for controlled delivery of biomacromolecules or even living cells. Given these unique properties of the dynamic covalent bonds, DDSs formulated with the dynamic covalent bonds can be rationally designed for improved cancer therapy by promoting tumor penetration, increasing the cellular uptake and facilitating the payload release inside the tumor cells.
|
|
Table 1 The summary of dynamic covalent bonds-based DDSs. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Despite promising, there are still some challenges for the clinical translation of the dynamic covalent bond-based DDSs. As the physiological environment is complicated, low tumor specificity and insufficient serum stability are two of the major obstacles for the development of dynamic covalent bond-based DDSs. Up to date, most of the stimuli-activatable DDSs are designed to be acid or GSH-responsive, which display unsatisfying tumor specificity since the acidic and reduction microenvironment are also shared by the normal tissues/cells. Meanwhile, the dynamic feature of some dynamic covalent bonds also impairs the serum stability of the DDSs. For example, the boronic ester-based DDSs can be exchanged with blood glucose. Therefore, it is of pivotal priority to develop novel kinds of dynamic covalent bonds with improved tumor specificity and serum stability for clinical translation of the dynamic covalent bond-based DDSs.
Declaration of competing interestThe authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
AcknowledgmentsFinancial supports from the National Natural Science Foundation of China (Nos. 31671024, 51873228 and 31622025), the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2014218), the Fusion Grant between Fudan University and Shanghai Institute of Materia Medica, CAS (No. FU-SIMM- 20182006), and the Open Project Program of Key Lab of Smart Drug Delivery (Ministry of Education), Department of Pharmaceutics, School of Pharmacy, Fudan University, China are gratefully acknowledged.
| [1] |
T.M. Allen, P.R. Cullis, Science 303 (2004) 1818-1822. DOI:10.1126/science.1095833 |
| [2] |
G.C. Wu, T. Zhao, D.W. Kang, et al., J. Med. Chem. 62 (2019) 9375-9414. DOI:10.1021/acs.jmedchem.9b00359 |
| [3] |
W.Q. Wang, M. Saeed, Y. Zhou, et al., J. Gene Med. 21 (2019) e3092. |
| [4] |
H.J. Yu, R.Q. Huang, Y. Nie, J. Gene Med. 21 (2019) e3111. |
| [5] |
H. Maeda, Adv. Drug Deliv. Rev. 91 (2015) 3-6. DOI:10.1016/j.addr.2015.01.002 |
| [6] |
M. Srinivasarao, P.S. Low, Chem. Rev. 117 (2017) 12133-12164. DOI:10.1021/acs.chemrev.7b00013 |
| [7] |
M. Srinivasarao, C.V. Galliford, P.S. Low, Nat. Rev. Drug Discov. 14 (2015) 203-219. DOI:10.1038/nrd4519 |
| [8] |
P.T. Wong, S.K. Choi, Chem. Rev. 115 (2015) 3388-3432. DOI:10.1021/cr5004634 |
| [9] |
J.M. Lambert, Mol. Pharm. 12 (2015) 1701-1702. DOI:10.1021/acs.molpharmaceut.5b00302 |
| [10] |
A. Mullard, Nat. Rev. Drug Discov. 12 (2013) 329-332. DOI:10.1038/nrd4009 |
| [11] |
S. Cazzamalli, A.D. Corso, F. Widmayer, D. Neri, J. Am. Chem. Soc. 140 (2018) 1617-1621. DOI:10.1021/jacs.7b13361 |
| [12] |
I. Ekladious, Y.L. Colson, M.W. Grinstaff, Nat. Rev. Drug Discov. 18 (2019) 273-294. DOI:10.1038/s41573-018-0005-0 |
| [13] |
S. Biswas, J. Das, S. Barman, et al., ACS Appl. Mater. Interfaces 9 (2017) 28180-28184. DOI:10.1021/acsami.7b05132 |
| [14] |
H. Wang, Q.W. Chen, S.Q. Zhou, Chem. Soc. Rev. 47 (2018) 4198-4232. DOI:10.1039/C7CS00399D |
| [15] |
J. Lu, F. Jiang, A.P. Lu, G. Zhang, Int. J. Mol. Sci. 17 (2016) 561. DOI:10.3390/ijms17040561 |
| [16] |
J.D. Bargh, A. Isido-Llobet, J.S. Parker, D.R. Spring, Chem. Soc. Rev. 48 (2019) 4361-4374. DOI:10.1039/C8CS00676H |
| [17] |
F. Schaufelberger, B.J.J. Timmer, O. Ramström, Principles of dynamic covalent chemistry, in: M. Zhang, Y.H. Jin (Eds.), Dynamic Covalent Chemistry: Principles, Reactions, and Applications, John Wiley & Sons, West Sussex, 2018, pp. 1-30.
|
| [18] |
T.A. Baillie, Angew. Chem. Int. Ed. 55 (2016) 13408-13421. DOI:10.1002/anie.201601091 |
| [19] |
T. Maeda, H. Otsuka, A. Takahara, Dynamic combinatorial methods in materials science, in: B.L. Miller (Ed.), Dynamic Covalent Chemistry: In Drug Dicovery, Bioorganic Chemistry and Materials Science, John Wiley & Sons, Hoboken, 2010, pp. 229-260.
|
| [20] |
A. Herrmann, Chem. Soc. Rev. 43 (2014) 1899-1933. DOI:10.1039/C3CS60336A |
| [21] |
Y. Liu, J.M. Lehn, A.K.H. Hirsch, Acc. Chem. Res. 50 (2017) 376-386. DOI:10.1021/acs.accounts.6b00594 |
| [22] |
S. Ulrich, Acc. Chem. Res. 52 (2019) 510-519. DOI:10.1021/acs.accounts.8b00591 |
| [23] |
R.J. Wojtecki, M.A. Meador, S.J. Rowan, Nat. Mater. 10 (2011) 14-27. DOI:10.1038/nmat2891 |
| [24] |
M. Bruneau, S. Bennici, J. Brendle, et al., J. Control. Release 294 (2019) 355-371. DOI:10.1016/j.jconrel.2018.12.038 |
| [25] |
R. Tang, M. Wang, M. Ray, et al., J. Am. Chem. Soc. 139 (2017) 8547-8551. DOI:10.1021/jacs.7b02801 |
| [26] |
W. Cao, L. Wang, H.P. Xu, Nano Today 10 (2015) 717-736. DOI:10.1016/j.nantod.2015.11.004 |
| [27] |
D. Konetski, S. Mavila, C. Wang, B. Worrell, C.N. Bowman, Chem. Commun. 54 (2018) 8108-8111. DOI:10.1039/C8CC03471K |
| [28] |
Y. Liu, B. Fan, Q. Shi, et al., ACS Nano 13 (2019) 6760-6769. DOI:10.1021/acsnano.9b01343 |
| [29] |
H.A. Houck, E. Blasco, F.E. du Prez, C.B. Kowollik, J. Am. Chem. Soc. 141 (2019) 12329-12337. DOI:10.1021/jacs.9b05092 |
| [30] |
J. Kida, D. Aoki, H. Otsuka, ACS Macro Lett. 8 (2018) 1-6. |
| [31] |
F. Amir, K.P. Liles, A. Delawder, et al., ACS Appl. Mater. Inter. 11 (2019) 24627-24638. DOI:10.1021/acsami.9b08853 |
| [32] |
S.L. Xiang, Q.X. Hua, W.L. Gong, et al., ACS Appl. Mater. Interfaces 11 (2019) 23623-23631. DOI:10.1021/acsami.9b06608 |
| [33] |
Z.T. Yang, Y. Peng, L.Y. Qiu, Chin. Chem. Lett. 29 (2018) 1839-1844. DOI:10.1016/j.cclet.2018.11.009 |
| [34] |
J.F. Reuther, A.C. Goodrich, P.R. Escamilla, et al., J. Am. Chem. Soc. 140 (2018) 3768-3774. DOI:10.1021/jacs.8b00046 |
| [35] |
T.N. Wang, G. Yang, L.B. Wu, G.S. Chen, Chin. Chem. Lett. 27 (2016) 1740-1744. DOI:10.1016/j.cclet.2016.05.009 |
| [36] |
X.L. Wu, C.L. He, Y.D. Wu, X.S. Chen, Biomaterials 75 (2016) 148-162. DOI:10.1016/j.biomaterials.2015.10.016 |
| [37] |
T.J. Dai, C.P. Wang, Y.Q. Wang, et al., ACS Appl. Mater. Interfaces 10 (2018) 15163-15173. DOI:10.1021/acsami.8b02527 |
| [38] |
J.J. Hu, Y.H. Chen, Y.Q. Li, Z.J. Zhou, Y.Y. Chen, Biomaterials 112 (2017) 133-140. DOI:10.1016/j.biomaterials.2016.10.015 |
| [39] |
W.D. Nie, W. Wei, L.P. Zuo, et al., ACS Nano 13 (2019) 1469-1478. DOI:10.1021/acsnano.8b07141 |
| [40] |
X.W. Guan, Z.P. Guo, L. Lin, et al., Nano Lett. 16 (2016) 6823-6831. DOI:10.1021/acs.nanolett.6b02536 |
| [41] |
X.X. Han, Y.Y. Li, Y. Xu, et al., Nat. Commun. 9 (2018) 3390. DOI:10.1038/s41467-018-05906-x |
| [42] |
J.J. Hu, Y.C. Quan, Y.P. Lai, et al., J. Control. Release 247 (2017) 145-152. DOI:10.1016/j.jconrel.2017.01.003 |
| [43] |
J.J. Hu, Z. Zheng, C.X. Liu, et al., Biomater. Sci. 7 (2019) 581-584. DOI:10.1039/C8BM01211C |
| [44] |
Z.Y. Li, F. Zhou, Z.Y. Li, et al., ACS Appl. Mater. Interfaces 10 (2018) 25194-25202. DOI:10.1021/acsami.8b08165 |
| [45] |
S.O. Doronina, B.E. Toki, M.Y. Torgov, et al., Nat. Biotechnol. 21 (2003) 778-784. DOI:10.1038/nbt832 |
| [46] |
H. Herrmann, S. Cerny-Reiterer, K.V. Gleixner, et al., Haematologica 97 (2012) 219-226. DOI:10.3324/haematol.2010.035006 |
| [47] |
I.R. Yurkiewicz, L. Muffly, M. Liedtke, Drug Des. Dev. Ther. 12 (2018) 2293-2300. DOI:10.2147/DDDT.S150317 |
| [48] |
O. Sedlacek, B.D. Monnery, J. Mattova, et al., Biomaterials 146 (2017) 1-12. DOI:10.1016/j.biomaterials.2017.09.003 |
| [49] |
A.K. Pearce, J.D. Simpson, N.L. Fletcher, et al., Biomaterials 141 (2017) 330-339. DOI:10.1016/j.biomaterials.2017.07.004 |
| [50] |
F. Roncato, F. Rruga, E. Porcù, et al., Nat. Commun. 9 (2018) 4070. DOI:10.1038/s41467-018-06602-6 |
| [51] |
H.J. Li, J.Z. Du, X.J. Du, et al., Proc. Natl. Acad. Sci. U. S. A. 113 (2016) 4164-4169. DOI:10.1073/pnas.1522080113 |
| [52] |
F.Y. Zhou, B. Feng, H.J. Yu, et al., Adv. Mater. 31 (2019) 1805888. DOI:10.1002/adma.201805888 |
| [53] |
A. Gao, X.L. Hu, M. Saeed, et al., Acta Pharmacol. Sin. 40 (2019) 1129-1137. DOI:10.1038/s41401-019-0281-1 |
| [54] |
M. Saeed, J. Gao, Y. Shi, T. Lammers, H. Yu, Theranostics 9 (2019) 7981-8000. DOI:10.7150/thno.37568 |
| [55] |
G.N. Grover, R.L. Braden, K.L. Christman, Adv. Mater. 25 (2013) 2937-2942. DOI:10.1002/adma.201205234 |
| [56] |
P.J. O'Brien, S. Elahipanah, D. Rogozhnikov, M.N. Yousaf, ACS Cent. Sci. 3 (2017) 489-500. DOI:10.1021/acscentsci.7b00132 |
| [57] |
B. Liu, S. Thayumanavan, J. Am. Chem. Soc. 139 (2017) 2306-2317. DOI:10.1021/jacs.6b11181 |
| [58] |
Y.Q. Tang, Z.Y. Zeng, X. He, et al., Adv. Sci. 4 (2017) 1600228. DOI:10.1002/advs.201600228 |
| [59] |
Y.N. Zhong, K. Goltsche, L. Cheng, et al., Biomaterials 84 (2016) 250-261. DOI:10.1016/j.biomaterials.2016.01.049 |
| [60] |
Y.D. Gu, Y.N. Zhong, F.H. Meng, et al., Biomacromolecules 14 (2013) 2772-2780. DOI:10.1021/bm400615n |
| [61] |
S.T. Guo, Y. Nakagawa, A. Barhoumi, et al., J. Am. Chem. Soc. 138 (2016) 6127-6130. DOI:10.1021/jacs.6b02435 |
| [62] |
E.M. Bachelder, T.T. Beaudette, K.E. Broaders, J. Dashe, J.M.J. Fréchet, J. Am. Chem. Soc. 130 (2008) 10494-10495. DOI:10.1021/ja803947s |
| [63] |
J.A. Cohen, T.T. Beaudette, J.L. Cohen, et al., Adv. Mater. 22 (2010) 3593-3597. DOI:10.1002/adma.201000307 |
| [64] |
L.N. Cui, J.L. Cohen, C.K. Chu, et al., J. Am. Chem. Soc. 134 (2012) 15840-15848. DOI:10.1021/ja305552u |
| [65] |
Y. Fu, W. Liu, L.Y. Wang, et al., Adv. Funct. Mater. 28 (2018) 1802250. DOI:10.1002/adfm.201802250 |
| [66] |
Y. Zou, Y.J. Liu, D.Y. Zhang, et al., Adv. Mater. 30 (2018) 1803717. DOI:10.1002/adma.201803717 |
| [67] |
A. Yurkovetskiy, S. Choi, A. Hiller, et al., Biomacromolecules 6 (2005) 2648-2658. DOI:10.1021/bm049210k |
| [68] |
V. Le Joncour, A. Martins, M. Puhka, et al., Mol. Cancer Ther. 18 (2019) 1721-1730. DOI:10.1158/1535-7163.MCT-19-0207 |
| [69] |
E. Kumazawa, Y. Ochi, Cancer Sci. 95 (2004) 168-175. DOI:10.1111/j.1349-7006.2004.tb03199.x |
| [70] |
A. Kulkarni, K. DeFrees, S. Hyun, et al., J. Am. Chem. Soc. 134 (2012) 7596-7599. DOI:10.1021/ja300690j |
| [71] |
Q. Zha, X. Wang, X. Cheng, et al., Mater. Sci. Eng. C-Mater. 78 (2017) 246-257. DOI:10.1016/j.msec.2017.04.098 |
| [72] |
L.P. Qiu, M.Q. Zhu, K. Gong, et al., Mater. Sci. Eng. C-Mater. 78 (2017) 912-922. DOI:10.1016/j.msec.2017.04.137 |
| [73] |
G.Q. Yan, J. Wang, P.P. Zhang, et al., Polym. Chem. 8 (2017) 2063-2073. DOI:10.1039/C6PY02204A |
| [74] |
S. Cambray, J. Gao, Acc. Chem. Res. 51 (2018) 2198-2206. DOI:10.1021/acs.accounts.8b00154 |
| [75] |
G.A. Ellis, M.J. Palte, R.T. Raines, J. Am. Chem. Soc. 134 (2012) 3631-3634. DOI:10.1021/ja210719s |
| [76] |
W.X. Chen, Y.F. Cheng, B.H. Wang, Angew. Chem. Int. Ed. 51 (2012) 5293-5295. DOI:10.1002/anie.201201179 |
| [77] |
Y.P. Li, W.W. Xiao, K. Xiao, et al., Angew. Chem. Int. Ed. 51 (2012) 2864-2869. DOI:10.1002/anie.201107144 |
| [78] |
Y. Ma, P.Y. He, X.H. Tian, et al., ACS Appl. Mater. Interfaces 11 (2019) 23948-23956. DOI:10.1021/acsami.9b09031 |
| [79] |
J.Y. Zhu, X. Zeng, S.Y. Qin, Biomaterials 83 (2016) 79-92. DOI:10.1016/j.biomaterials.2016.01.003 |
| [80] |
S.C. Tang, H. Ma, H.C. Tu, et al., Adv. Sci. 5 (2018) 1800638. DOI:10.1002/advs.201800638 |
| [81] |
W.X. Gao, Y.L. Hu, L. Xu, et al., Chin. Chem. Lett. 29 (2018) 1795-1798. DOI:10.1016/j.cclet.2018.05.022 |
| [82] |
N.A. Bakh, A.B. Cortinas, M.A. Weiss, et al., Nat. Chem. 9 (2017) 937-943. DOI:10.1038/nchem.2857 |
| [83] |
A. Matsumoto, M. Tanaka, H. Matsumoto, et al., Sci. Adv. 3 (2017) eaaq0723. DOI:10.1126/sciadv.aaq0723 |
| [84] |
C.Y. Liu, W.W. Shen, B.N. Li, et al., Chem. Mater. 31 (2019) 1956-1965. DOI:10.1021/acs.chemmater.8b04672 |
| [85] |
X.J. Cheng, M.Y. Li, H. Wang, Y.Y. Cheng, Chin. Chem. Lett. 31 (2020) 869-874. DOI:10.1016/j.cclet.2019.07.013 |
| [86] |
S.C. Liu, J.M. Pan, J.X. Liu, et al., Small 14 (2018) 1703968. DOI:10.1002/smll.201703968 |
| [87] |
C.P. Wang, H.J. S, Y.T. Wang, et al., Nano Lett. 18 (2018) 7045-7051. DOI:10.1021/acs.nanolett.8b03015 |
| [88] |
L. Zhang, Y.M. Zhang, G.X. Liu, Y. Liu, Chin. Chem. Lett. 30 (2019) 120-122. DOI:10.1016/j.cclet.2018.04.028 |
| [89] |
S. Liu, Y.S. Gao, D.Z. Zhou, et al., Nat. Commun. 10 (2019) 3307. DOI:10.1038/s41467-019-11190-0 |
| [90] |
X.Y. Jiang, B.J. Du, J. Zheng, Nat. Nanotechnol. 14 (2019) 874-882. DOI:10.1038/s41565-019-0499-6 |
| [91] |
X.Y. Tan, X.G. Lu, F. Jia, et al., J. Am. Chem. Soc. 138 (2016) 10834-10837. DOI:10.1021/jacs.6b07554 |
| [92] |
Q. Zhou, S.Q. Shao, J.Q. Wang, et al., Nat. Nanotechnol. 14 (2019) 799-809. DOI:10.1038/s41565-019-0485-z |
| [93] |
Y. Qu, B.Y. Chu, X.W. Wei, et al., J. Control. Release 296 (2019) 93-106. DOI:10.1016/j.jconrel.2019.01.016 |
| [94] |
R.V.J. Chari, ACS Med. Chem. Lett. 7 (2016) 974-976. DOI:10.1021/acsmedchemlett.6b00312 |
| [95] |
R.G. Qi, Y.H. Wang, P.M. Bruno, et al., Nat. Commun. 8 (2017) 2166. DOI:10.1038/s41467-017-02390-7 |
| [96] |
B. Feng, B. Hou, Z.A. Xu, et al., Adv. Mater. (2019) 1902960. |
| [97] |
P.Y. Yuan, H.L. Zhang, L.H. Qian, et al., Angew. Chem. Int. Ed. 56 (2017) 12481-12485. DOI:10.1002/anie.201705578 |
| [98] |
X. Ling, X. Chen, I.A. Riddell, et al., Nano Lett. 18 (2018) 4618-4625. DOI:10.1021/acs.nanolett.8b01924 |
| [99] |
H.P. Xu, W. Cao, X. Zhang, Acc. Chem. Res. 46 (2013) 1647-1658. DOI:10.1021/ar4000339 |
| [100] |
B.J. Sun, C. Luo, X.B. Zhang, et al., Nat. Commun. 10 (2019) 3211. DOI:10.1038/s41467-019-11193-x |
| [101] |
Y. Li, Y.H. Li, W.H. Ji, et al., J. Am. Chem. Soc. 140 (2018) 4164-4171. DOI:10.1021/jacs.8b01641 |
| [102] |
D. Shao, M.Q. Li, Z. Wang, et al., Adv. Mater. 30 (2018) 1801198. DOI:10.1002/adma.201801198 |
| [103] |
B. Feng, F.Y. Zhou, B. Hou, et al., Adv. Mater. 30 (2018) 1803001. DOI:10.1002/adma.201803001 |
| [104] |
X.D. Xue, Y. Huang, R.N. Bo, et al., Nat. Commun. 9 (2018) 3653. DOI:10.1038/s41467-018-06093-5 |
| [105] |
Y.Y. He, G. Cheng, L. Xie, et al., Adv. Mater. 26 (2014) 1534-1540. DOI:10.1002/adma.201304592 |