 2020, Vol. 31
2020, Vol. 31 


b College of Chemistry, Zhengzhou University, Zhengzhou 450001, China;
c School of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Theoretical and Computational Chemistry, Chongqing University, Chongqing 4000030, China
Asymmetric construction of C-C bonds is one of the hottest topics in organic chemistry as a way to rapidly access stereoenriched skeletons. Within this area, the benzoin reaction, which was first reported by Liebig and coworkers in 1832 [1], is one of the most useful approaches for. With the rapid development of organocatalysis, the use of N-heterocyclic carbenes (NHCs) for asymmetric construction of carbon-carbon and carbonheteroatom bonds has attracted increasing attention during the past decades [2-8]. Importantly, NHC catalysts have the unique property of polarity inversion, and concurrently offer alternatives for governing stereo-, chemo-, and regioselectivity [9-22]. As a result, the NHC-catalyzed asymmetric construction of C-C bonds has been developed into a valuable approach in organic synthesis.
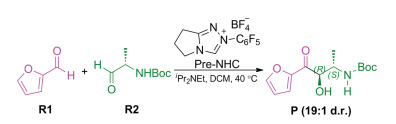
As an important example, NHC-catalyzed cross-benzoin reactions have exhibited remarkable utility in a wide variety of reactions reported in recent years [23-27]. Recent studies have achieved high enantioselectivity in homo-coupling reactions; however, the cross-coupling reaction between two different aldehydes is still challenging because there are four different coupling modes: two homo-couplings and two cross-couplings. Some studies have explored the origin of stereoselectivity in chiral NHC catalyzed systems, but the use of chiral substrates for chirality induction, further leading to the desired diastereoselectivity, is unexplored to date. As shown in Scheme 1, Gravel and coworkers reported an N-heterocyclic carbene-catalyzed cross-benzoin reaction with excellent diastereoselectivity by using a chiral substrate [28]. To the best of our knowledge, the origin of this diastereoselectivity is still unclear, through it would be important knowledge for rational design. Herein, DFT, which has proved one of the most powerful and efficient tools for uncovering the mechanisms and predicting the selectivities of organic reactions enabled by organocatalysts [29-35] and transition metal catalysts [36-51], was employed to disclose the origin of the diastereoselectivity.

|
Download:
|
| Scheme 1. Selected model employed in the calculations. | |
{kind=link}
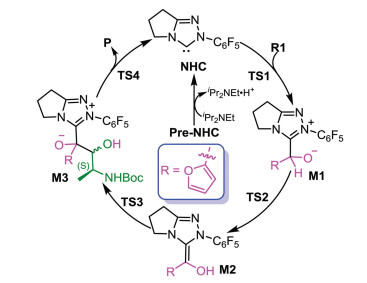
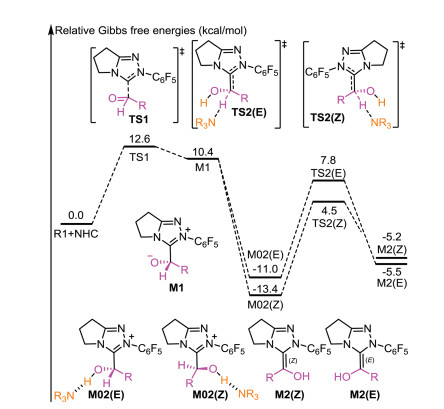
As shown in Scheme 2, a possible mechanism of the title reaction was suggested based on the experimental details. Initially, the reaction started with the nucleophilic addition of NHC onto furfural (R1) to give zwitterionic intermediate M1 via transition state TS1. The energy barrier of this process was calculated to be 12.6 kcal/mol relative to separated R1 and NHC catalyst (Fig. 1). The next step is the formation of a Breslow intermediate through an intramolecular [1, 2]-proton transfer process, in which two configured (E and Z) Breslow intermediate would be formed. Previous computational studies had confirmed that protic media play an important role in such proton transfer processes [52-54]. Noteworthy, the formation of the active NHC catalyst and Brønsted acid iPr2NEt·H+ first occurs via abstracting the proton of the original Pre-NHC by iPr2NEt. Thus, the in-situ-generated Brønsted acid iPr2NEt·H+, used as the protic medium, was considered when calculating the proton transfer barriers. As shown in Fig. 1, the energy barriers required for this transformation were 17.9 and 18.8 kcal/mol for transition states T\S2(Z) and TS2(E), respectively. With regard to the selectivity, these two pathways should be competing processes under the experimental conditions.

|
Download:
|
| Scheme 2. Possible mechanisms of the NHC-catalyzed cross-benzoin reaction. | |
{kind=link}

|
Download:
|
| Fig. 1. Energy profile for the formation of E/Z-type Breslow intermediate. | |
{kind=link}
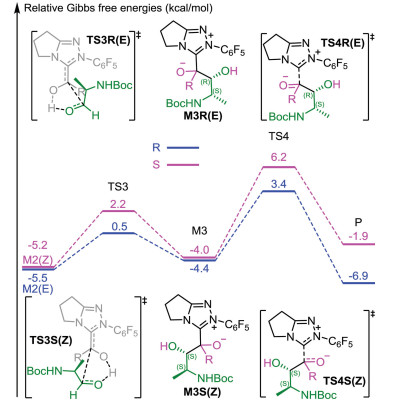
Then, an intermolecular coupling between the Breslow intermediate and an α-amino aldehyde (R2) led to the intermediate M3 through the transition state TS3, and the chiral center appeared during the C-C bond formation process. The computational results indicated that the formation of the C-C bond was accompanied by proton transfer. To confirm the reliability of the calculated results, both coupling modes were calculated to verify the lowest energy conformers of transition states involved in this step (the computational results are provided in Fig. S1 in Supporting Information) and only the lowest energy conformers were used in the following discussion. As shown in Fig. 2, the transition structure leading to the R-product ((E)-TS3R) was calculated to be 1.4 kcal/mol more stable than that leading to the S-product ((Z)-TS3S). This revealed that the R-configured pathway was more energetically favorable than the S-configured pathway and the computational results align well with experimental observations. Next, the dissociation of NHC with generation of the final product happened via transition state TS4. The energy barriers of this step were 7.8 and 10.2 kcal/mol for transition states (E)-TS4R and (Z)-TS4S respectively (Fig. 2), showing that the NHC catalyst was easy to regenerate. As for the previous step, by comparing the energy barriers of the R- and S-configured pathways, one can conclude that the former is more energetically favorable. All the calculations are consistent with the experimental observations.

|
Download:
|
| Fig. 2. Energy profile of diastereoselective coupling process. | |
{kind=link}
Having established the most energetically favorable pathway, we then turned our attention to the origin of diastereoselectivity. As discussed above, the C-C bond formation process was identified to be the diastereoselectivity-determining step. The total energy barriers of the reaction are 17.9/18.8 kcal/mol for forming Z/E-type Breslow intermediate, which is competing pathways. To gain insight into the diastereoselectivity, the key transition structures involved in the C-C bond formation were analyzed. As sated above, the transition state leading to the R-configured product was predicted to be 1.4 kcal/mol more stable than that leading to the S-configured product. The value of diastereomeric excess was predicted to be 83%, which was in agreement with experimental observations.
Then, non-covalent interaction (NCI) analysis, which has been successfully used to identify electrostatic interactions, was performed by using the Multiwfn software [55]. As shown in Fig. 3, the key geometric features of TS3R(E) and TS3S(Z) were mapped, in which the attractive and repulsive interactions are colored green and red, respectively. C-H…O interactions existed in both transition states TS3R(E) and TS3S(Z). This type of interactions made nearly equal contributions in both transition states. However, some distinct C-H…F interactions were found only in TS3R(E). These additional interactions were responsible for lowering the energy of the R-configured transition state. Overall, the origin of the diastereoselectivity was determined by stronger hydrogen bond interactions in TS3R(E) (C-H…O and C-H…F).

|
Download:
|
| Fig. 3. NCI analysis for the diastereocontrolling transition states TS3R(E) and TS3S (Z). Values in parentheses are distance between two interacting fragments. | |
{kind=link}
In summary, the detailed reaction mechanism and origin of the diastereoselectivity of NHC-catalyzed cross-benzoin reaction were investigated by the M06-2X method, the computational details are provided in Supporting information. Based on the computational results, the coupling reaction of the α-amino aldehyde and Breslow intermediate was identified as the diastereoselectivity-determining step. The stronger hydrogen bonds betweenthe twointeracting fragments in the R-configured pathway were responsible for controllingthe diastereoselectivity, which were further recognized as C-H…O and C-H…F interactions by NCI analysis.
AcknowledgmentThe authors gratefully thank the financial support from the National Science Foundation of China (Nos. 21822303 and 21772020) and Startup Fund of Zhengzhou University of Light Industry (No. 2017BSJJ036).
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.010.
| [1] |
F. Wöhler, J. Liebig, Ann. Pharm. 3 (1832) 249-282. DOI:10.1002/jlac.18320030302 |
| [2] |
K.J.R. Murauski, A.A. Jaworski, K.A. Scheidt, Chem.Soc.Rev. 47 (2018) 1773-1782. DOI:10.1039/C7CS00386B |
| [3] |
S. Mukherjee, A.T. Biju, Chem. -Asian J. 13 (2018) 2333-2349. DOI:10.1002/asia.201800902 |
| [4] |
R.S. Akkattu, A.T. Biju, V. Nair, Chem. Soc. Rev. 44 (2015) 5040-5052. DOI:10.1039/C5CS00162E |
| [5] |
Y. Wang, W.J. Zhang, D.H. Wei, ChemCatChem 10 (2018) 338-360. DOI:10.1002/cctc.201701119 |
| [6] |
X.Y. Chen, S. Li, F. Verica, M. Kumar, D. Enders, iScience 2 (2018) 1-26. |
| [7] |
D.M. Flanigan, F. Romanov-Michailidis, N.A. White, T. Rovis, Chem. Rev. 115 (2015) 9307-9387. DOI:10.1021/acs.chemrev.5b00060 |
| [8] |
X.Y. Chen, Q. Liu, P. Chauhan, D. Enders, Angew. Chem. Int. Ed. 57 (2018) 3862-3873. DOI:10.1002/anie.201709684 |
| [9] |
P.C. Tu, L. Zhou, A.M. Kirillov, R. Fang, L.Z. Yang, Org. Chem. Front. 5 (2018) 1356-1365. DOI:10.1039/C8QO00139A |
| [10] |
Y. Wang, Y.Y. Wang, X.H. Wang, et al., Catal. Sci. Technol. 8 (2018) 4229-4240. DOI:10.1039/C8CY01272E |
| [11] |
X.X. Wei, R. Fang, L.Z. Yang, Catal. Sci. Technol. 5 (2015) 3352-3362. DOI:10.1039/C5CY00407A |
| [12] |
X. Li, S.J. Li, Y.Y. Wang, et al., Catal. Sci. Technol. 9 (2019) 2514-2522. DOI:10.1039/C9CY00526A |
| [13] |
Y. Wang, D.H. Wei, M.S. Tang, J. Org. Chem. 82 (2017) 13043-13050. DOI:10.1021/acs.joc.7b01992 |
| [14] |
Y. Wang, Y. Qiao, D.H. Wei, M.S. Tang, Org. Chem. Front. 4 (2017) 1987-1998. DOI:10.1039/C7QO00436B |
| [15] |
T. Liu, S.M. Han, Y.P. Li, S.W. Bi, J. Org. Chem. 81 (2016) 9775-9784. DOI:10.1021/acs.joc.6b01838 |
| [16] |
Q.Q. Shi, Y. Wang, Y. Wang, et al., Org. Chem. Front. 5 (2018) 2739-2748. DOI:10.1039/C8QO00716K |
| [17] |
X. Li, Y.Y. Wang, Y. Wang, et al., J. Org. Chem. 83 (2018) 8543-8555. DOI:10.1021/acs.joc.8b01112 |
| [18] |
Y. Wang, S.R. Zhang, Y.Y. Wang, L.B. Qu, D.H. Wei, Org. Chem. Front. 5 (2018) 2065-2072. DOI:10.1039/C8QO00398J |
| [19] |
Y. Wang, L.B. Qu, Y. Lan, D.H. Wei, ChemCatChem 11 (2019) 2919-2925. DOI:10.1002/cctc.201900424 |
| [20] |
Y. Wang, Q.Y. Wu, T.H. Lai, et al., Catal. Sci. Technol. 9 (2019) 465-476. DOI:10.1039/C8CY02238K |
| [21] |
Y. Wang, L.B. Qu, D.H. Wei, Chem. -Asian J. 14 (2019) 293-30014. |
| [22] |
B.B. Hu, T.X. Liu, P.L. Zhang, et al., Org. Lett. 20 (2018) 4801-4805. DOI:10.1021/acs.orglett.8b01956 |
| [23] |
Y. Ma, S. Wei, J. Wu, et al., Adv. Synth. Catal. 350 (2008) 2645-2651. DOI:10.1002/adsc.200800371 |
| [24] |
S.M. Langdon, M.M.D. Wilde, K. Thai, M. Gravel, J. Am. Chem. Soc. 136 (2014) 7539-7542. DOI:10.1021/ja501772m |
| [25] |
S.M. Langdon, C.Y. Legault, M. Gravel, J. Org. Chem. 80 (2015) 3597-3610. DOI:10.1021/acs.joc.5b00301 |
| [26] |
P. Haghshenas, J.W. Quail, M. Gravel, J. Org. Chem. 81 (2016) 12075-12083. DOI:10.1021/acs.joc.6b02568 |
| [27] |
W. Zhang, Y. Wang, D.H. Wei, M.S. Tang, X.J. Zhu, Org. Biomol. Chem. 14 (2016) 6577-6590. DOI:10.1039/C6OB00791K |
| [28] |
P. Haghshenas, M. Gravel, Org. Lett. 18 (2016) 4518-4521. DOI:10.1021/acs.orglett.6b02123 |
| [29] |
J.Y. Zhang, Y.X. Shao, Y.W. Li, Y. Liu, Z.F. Ke, Chin. Chem. Lett. 29 (2018) 1226-1232. DOI:10.1016/j.cclet.2018.02.007 |
| [30] |
H.H. Chen, L. Zhu, K.B. Zhong, et al., Chin. Chem. Lett. 29 (2018) 1237-1241. DOI:10.1016/j.cclet.2018.03.018 |
| [31] |
H. Li, X. Hong, Chin. Chem. Lett. 29 (2018) 1585-1590. DOI:10.1016/j.cclet.2018.01.030 |
| [32] |
W.J. Yao, Z.Y. Yu, S. Wen, et al., Chem. Sci. 8 (2017) 5196-5200. DOI:10.1039/C7SC00952F |
| [33] |
H.M. Zhang, H. Xu, H.N. Bai, et al., Org. Chem. Front. 5 (2018) 1493-1501. DOI:10.1039/C8QO00129D |
| [34] |
H.Z. Ni, Z.Y. Yu, W.J. Yao, et al., Chem. Sci. 8 (2017) 5699-5704. DOI:10.1039/C7SC02176C |
| [35] |
C.X. Cui, C.H. Shan, Y.P. Zhang, et al., Chem. -Asian J. 13 (2018) 1076-1088. DOI:10.1002/asia.201800146 |
| [36] |
S.W. Bi, P. Liu, B.P. Ling, X.G. Yuan, Y.Y. Jiang, Chin. Chem. Lett. 29 (2018) 1264-1268. DOI:10.1016/j.cclet.2017.11.043 |
| [37] |
L.B. Xu, H.H. Chen, J. Liu, et al., Org. Chem. Front. 6 (2019) 1162-1167. |
| [38] |
Y.Z. Li, H.H. Chen, L.B. Qu, K.N. Houk, Y. Lan, ACS Catal. 9 (2019) 7154-7165. DOI:10.1021/acscatal.9b02085 |
| [39] |
X.N. Ke, C.M. Schienebeck, C.C. Zhou, X.F. Xu, W.P. Tang, Chin. Chem. Lett. 26 (2015) 730-734. DOI:10.1016/j.cclet.2015.03.016 |
| [40] |
L. Zhou, L. Yang, Y.W. Zhang, et al., Org. Chem. Front. 6 (2019) 2701-2712. DOI:10.1039/C9QO00534J |
| [41] |
Z.Y. Wang, L. Zhu, K.B. Zhong, et al., ChemCatChem 10 (2018) 5280-5286. DOI:10.1002/cctc.201801301 |
| [42] |
Y. Wang, C. Du, Y.Y. Wang, et al., Adv. Synth. Catal. 360 (2018) 2668-2677. DOI:10.1002/adsc.201800036 |
| [43] |
Y. Qiao, J.M. Zhao, J.B. Chang, D.H. Wei, ChemCatChem 11 (2019) 780-789. DOI:10.1002/cctc.201801531 |
| [44] |
B. Lian, L. Zhang, D.C. Fang, Org. Chem. Front. 6 (2019) 2600-2606. DOI:10.1039/C9QO00154A |
| [45] |
C.H. Shan, K.B. Zhong, X.T. Qi, et al., Org. Chem. Front. 5 (2018) 3178-3185. DOI:10.1039/C8QO00699G |
| [46] |
H.N. Bai, H. Xu, H.M. Zhang, et al., Catal. Sci. Technol. 8 (2018) 5165-5177. DOI:10.1039/C8CY01322E |
| [47] |
H.N. Bai, H.M. Zhang, X.C. Wang, et al., J. Org. Chem. 84 (2019) 6709-6718. DOI:10.1021/acs.joc.9b00329 |
| [48] |
L. Zhu, Z.Y. Wang, S. Liu, et al., Chin. Chem. Lett. 30 (2019) 889-894. DOI:10.1016/j.cclet.2019.03.024 |
| [49] |
H.Y. Zou, Z.L. Wang, Y. Cao, G.P. Huang, Chin. Chem. Lett. 29 (2018) 1355-1358. DOI:10.1016/j.cclet.2017.10.034 |
| [50] |
Q. Xiong, D.D. Xu, C.H. Shan, et al., Chem. -Asian J. 14 (2019) 655-661. DOI:10.1002/asia.201801862 |
| [51] |
Z.R. Wang, P.P. Xie, Y.Z. Xia, Chin. Chem. Lett. 29 (2018) 47-53. DOI:10.1016/j.cclet.2017.06.018 |
| [52] |
Y. Wang, M.S. Tang, Y.Y. Wang, D.H. Wei, J. Org. Chem. 81 (2016) 5370-5380. DOI:10.1021/acs.joc.6b00656 |
| [53] |
Y.Y. Wang, D.H. Wei, Y. Wang, W.J. Zhang, M.S. Tang, ACS Catal. 6 (2016) 279-289. DOI:10.1021/acscatal.5b01710 |
| [54] |
Y. Wang, D.H. Wei, W.J. Zhang, et al., Org. Biomol. Chem. 12 (2014) 7503-7514. DOI:10.1039/C4OB01015A |
| [55] |
T. Lu, F.W. Chen, J. Comput. Chem. 33 (2012) 580-592. DOI:10.1002/jcc.22885 |