 2020, Vol. 31
2020, Vol. 31 


b Department of Medicinal Chemistry, Beijing Key Laboratory of Active Substances Discovery and Drugability Evaluation, Institute of Materia Medica, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing 100050, China
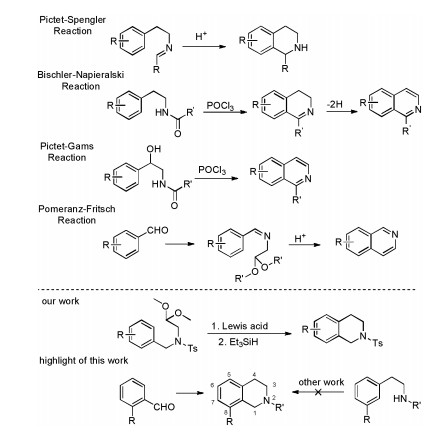
Tetrahydroisoquinoline (THIQ) is an important structural motif found in many alkaloid natural products [1] and several drugs including nomifensine [2], hydrastinine [3], dehydroemetine [4] and ecteinascidin 743 [5]. Beyond the classic PictetSpengler reaction and selective hydrogenation of isoquinolines, there are few reactions that can be used to synthesize THIQ conveniently [6]. The Pictet-Spengler reaction (Scheme 1) involves condensation of phenethylamines with an aldehyde or ketone to form an intermediate iminium ion that undergoes ring closure by electrophilic aromatic substitution, which limits the substrate scope to electron-rich aromatic systems. A Br ønsted acid catalyst in protic solvent is typically employed with heating thereby restricting the chemoselectivity. Because of the mechanism, meta-substituted phenethylamine analogues tend to afford 6-substituted THIQ over the 8-substituted THIQ regioisomers [7]. The related Bischler-Napieralski and PictetGams cyclization of phenylethylamides suffer from these same limitations. Complimentary to the above methods, the Pomeranz-Fritsch reaction is another strategy for the preparation of isoquinolines, especially 8-substituted THIQs. The reaction involves the acid-promoted condensation of a benzaldehyde with a 2, 2-dialkoxyethylamine, but is also restricted to electronrich benzaldehyde derivatives. Besides, other synthesis routes of THIQ derivatives such as visible-light-promoted transitionmetal-free approach [8] and silver-catalyzed radical cascade cyclization have been reported [9].

|
Download:
|
| Scheme 1. Methods for synthesis of THIQs and isoquinolines. | |
Here, we describe the serendipitous discovery of a one-pot route for the synthesis of THIQs via TiCl4 mediated ring-closure of benzylaminoacetal derivatives (Table 1).
|
|
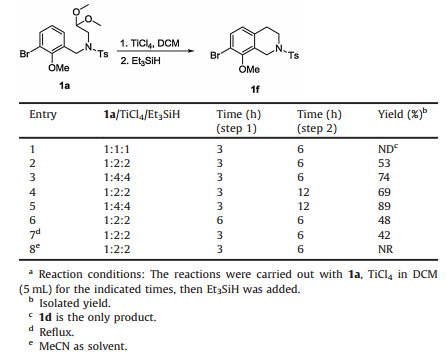
Table 1 Selected results for screening the reaction conditions.a |

Our initial goal was to synthesize THIQ analogue 1f through a Jackson-modifed Pomerantz-Fritsch reaction of 1a to afford 1b followed by hydrogenation [10]. Unfortunately, the desired product 1b was not observed using 6 mol/L aqueous HCl in dioxane and only 1c arising from acetal hydrolysis under the aqueous conditions was obtained in 85% yield. We next explored non-aqueous condition employing the Lewis acid AlCl3 in dichloromethane (DCM) and were heartened to observe 1b about 10% yield (just LC–MS confirmed).
Subsequently, we screened several Lewis acids to promote the cyclization of 1a to 1b including BF3·OEt2 [11], Bi(OTf)3, InCl3 and Al (OTf)3 in DCM at room temperature, but all exclusively furnished 1c (Table 1, entries 3–6). The substrate 1a decomposed with Eaton's reagent (Table 1, entry 7) and there was no reaction with SmI2 (Table 1, entry 8). To our surprise, treatment of 1a with TiCl4 in DCM at room temperature afforded 7-bromo-8-methoxy-2-tosyl- 1, 2, 3, 4-tetrahydroisoquinolin-4-ol (1d) in an impressive 85% yield (Table 1, entry 9). Based on our previous experience with reduction of benzyl ketones and alcohols with TiCl4/Et3SiH, we hypothesized addition of Et3SiH to the reaction mixture would directly afford the desired THIQ 1f [12]. We thus treated 1a with TiCl4 and Et3SiH in one pot, but observed only the formation of 1e (Table 1, entry 10). This results clearly indicates reduction proceeds faster than cyclization. We then tried a telescoping synthesis by the addition of TiCl4 to 1a in DCM followed by Et3SiH (1.5 mmol) after 3 h without any work-up and obtained 1f in 34% yield (Table 1, entry 11).
Encouraged by this result, we optimized conditions to obtain more satisfactory results (Table 2). To improve the applicability of the reaction, we examined other parameters including the ratio of reagents and the reaction time for each step. 1 equiv. of TiCl4 and Et3SiH produced only 1d instead of 1f (Table 2, entry 1). Increasing the amounts of TiCl4 and Et3SiH was necessary to promote the reaction, 2 equiv. of TiCl4/Et3SiH affording 1f in 53% yield, while 4 equiv. gave 1f in 74% yield (Table 2, entries 2 and 3). Prolonging the reaction time in step 2 from 6 h to 12 h led to higher yields, whereas increasing the reaction time of step one from 3 h to 6 h slightly diminished the yield (Table 2, entries 4–6). We explored other solvents (MeCN) as well as heating the reaction at reflux, but both modifications to the reaction conditions were deleterious (Table 2, entries 7 and 8). We also tried different N-protecting group such as N-acetyl, N-trifluoroacetyl and naked NH compounds as substrates. Unfortunately, only Ts group in the TiCl4 in DCM conditions works to afford corresponding product. Therefore, in this work Ts may be the most suitable group protects the Natom.
|
|
Table 2 Optimization of reaction conditions.a |
{kind=link}
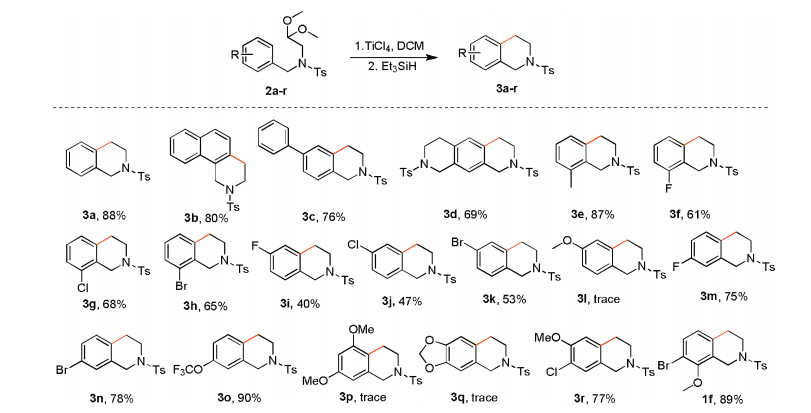
With the optimized reaction conditions (Table 2, entry 5), we examined the scope and limitation of this two-step, one-pot reaction for the synthesis of THIQs. As shown in Scheme 2, most of substrates (2a-r) afforded the desired products in good-toexcellent yields. To our delight, electron-withdrawing substituents were well tolerated with analogues substituted at the meta-position (3m and 3n) affording the highest overall yield followed in relative order by the ortho-position (3f-h) and para-position (3i-k). Exceeding our expectations, the meta-substituted substrates reacted with high regioselectivity to afford 3m-o. A clear limitation to the method is substrates bearing monomethoxy (2i) or dimethoxy (2p and 2q) groups, which did not react. It is noteworthy that introduction of an adjacent halogen substituent next to the alkoxy in 1a and 2r or replacement of the methyl ether with a trifluoromethyl ether in 2o provided 1f, 3r and 3o in the highest overall yields. The procedure for the synthesis and spectra data were shown in Supporting information. In these cases, substrates with strong electron-withdraw groups, such as -NO2, -CF3 could not react. Electron-donating group like -Me can afford the product with high yield. However, both mono-methoxy and dimethoxy compounds did not give products. We speculated that the lone pair electron of methoxy oxygen may coordinate with TiCl4 to form a strong electro-withdrawing effect, thereby deactivating the aromatic system. When a bromo- or chlorosubstituent is inserted next to MeO-, it blockades the formation of the methoxy-TiCl4 complex, thus the reaction works again. The advantage of this method could be used with weak electronwithdrawing group like halogens and -OCF3.

|
Download:
|
| Scheme 2. Substrate scope with variation of THIQ. All reactions werecarried out with 1a (1.0 mmol), TiCl4 (4.0 mmol) in DCM (5 mL) at room temperature for 3 h followed by the addition of Et3SiH (4.0 mmol) and stirring for another 12 h. Isolated yield. | |
{kind=link}
To explore the reaction process on a larger scale, we attempted the gram scale synthesis of 1f. We added TiCl4 (6.4 mL) in 1a (13.4 g) with 100 mL of DCM. Here control of the reaction temperature during the addition of TiCl4 wascritical toprevent a largeexotherm that compromised the yield and led to multiple byproducts. This was accomplished by slow addition and pre-cooling the reaction to -10 ℃. Finally, afforded 9.8 g 1f in 85% yield.
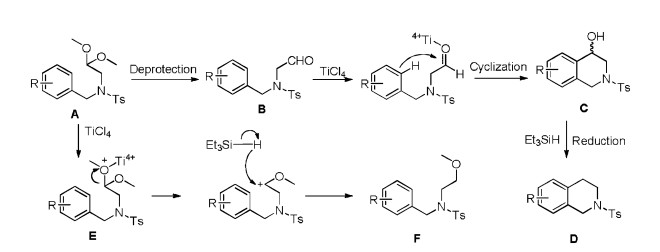
Actually, the proposed mechanism was basically disclosed when we screened the reaction condition. Scheme 3 describes the mechanism for the formation of compounds D and F from acetal A. Initially, acetal A is deprotected to furnish intermediate aldehyde B which can be isolated. In the absence of Et3SiH, B cyclizes to produce tetrahydroisoquinolin-4-ol C by TiCl4, which can also be isolated as the only product, but undergoes in situ reduction by Et3SiH to afford D in a telescoped reaction sequence. Compound D can be afforded from intermediate B and C. On the other hand, when the Et3SiH and TiCl4 are introduced simultaneously, the silane reacts directly with the oxocarbenium intermediate E to generate the undesired reduction product F.

|
Download:
|
| Scheme 3. The plausible mechanism. | |
{kind=link}
In conclusion, we have reported a mild and efficient method for the preparation of THIQs from benzylaminoacetal derivatives. The method employs TiCl4 to promote acetal deprotection and subsequent cyclization in DCM at room temperature, thereby avoiding high temperature as well as the strong Br ønsted acid employed in the traditional Pomeranz-Fritsch reaction. The telescoped reaction sequence uses Et3SiH in a second step to reduce the intermediate hydroxyl-THIQ to directly afford a THIQ. The method is noteworthy for its wide-substrate tolerance, accommodation of electron-withdrawing substituents in the aryl ring, and ability to give 8- substituent THIQs products that are not easily accessed by conventional approaches to THIQs. The combination of TiCl4/Et3SiH provides a useful and complementary method for the synthesis of valuable THIQ products from simple benzaldehyde derivatives.
AcknowledgmentsWe are grateful for financial support from Drug Innovation Major Project (No. 2018ZX09711-001-005) and CAMS Collaborative Innovation Project (No. 2017-I2M-2-004).
Appendix A. Supplementary dataSupplementary material related to this article canbe found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.09.023.
| [1] |
(a) J.D. Scott, R.M. Williams, Chem. Rev. 102 (2002) 1669-1730; (b) Y. Kashiwasa, A. Aoshima, Y. Ikeshiro, et al., Bioorg. Med. Chem. 13 (2005) 443-448. |
| [2] |
(a) A.D. Pechulis, J.P. Beck, M.A. Curry, et al., Bioorg. Med. Chem. Lett. 22 (2012) 7219-7222; (b) W.O. Shekim, A. Masterson, D.P. Cantwell, et al., J. Nerv. Ment. Dis. 177 (1989) 296-299; (c) E. Zara-Kaczian, L. Gyorgy, G. Deak, et al., J. Med. Chem. 29 (1986) 1189-1195. |
| [3] |
R.W. Freudenmann, F. Oxler, S. Bernschneider-Reif, Addiction 101 (2006) 1241-1245. DOI:10.1111/j.1360-0443.2006.01511.x |
| [4] |
G.H. Al-Khateeb, T.I. Al-Jeboori, K.A. Al-Janabi, Chemotherapy 23 (1977) 267-275. |
| [5] |
E.J. Corey, D.Y. Gin, R.S. Kania, J. Am. Chem. Soc. 118 (1996) 9202-9203. DOI:10.1021/ja962480t |
| [6] |
(a) A. Pictet, T. Spengler, Ber. Dtsch. Chem. Ges. 44 (1911) 2030-2036; (b) S.W. Youn, J. Org. Chem. 71 (2006) 2521-2523. |
| [7] |
R. Quevedo, E. Baquero, M. Rodriguez, Tetrahedron Lett. 51 (2010) 1774-1778. DOI:10.1016/j.tetlet.2010.01.115 |
| [8] |
X.C. Liu, K. Sun, X.L. Chen, et al., Adv. Synth. Catal. 361 (2019) 3712-3717. DOI:10.1002/adsc.201900544 |
| [9] |
K. Sun, Y.F. Si, X.L. Chen, et al., Adv. Synth. Catal. 361 (2019) 4483-4488. DOI:10.1002/adsc.201900691 |
| [10] |
(a) A.J. Birch, A.H. Jackson, P.V.R. Shannon, J. Chem. Soc. Perkin Trans. 1 (1974) 2185-2190; (b) D.L. Boger, C.E. Brotherton, M.D. Kelley, Tetrahedron 37 (1981) 3977-3980. |
| [11] |
M.J.E. Hewlins, R. Salter, Synth. Stuttg. (2007) 2157-2163. |
| [12] |
S. Bhattacharyya, J. Org. Chem. 63 (1998) 7101-7102. DOI:10.1021/jo980685+ |