 2020, Vol. 31
2020, Vol. 31 


α-Acyloxycarbonyl compounds are significant building blocks in synthetic organic chemistry [1] that can readily transform into various other functional groups, such as imidazoles, furans and oxazoles [2]. Traditionally, α-acyloxycarbonyl compounds can be prepared by the substitution reaction between α-halocarbonyl compounds [3] and α-diazoketones [4] with alkaline carboxylates or by the direct oxidative coupling of carbonyl compounds with toxic heavy metal oxidants suchas Pb(OAc)4, Tl(OAc)3andMn(OAc)3 [5]. Cu-catalysed oxidative coupling of ketones and carboxylic acids with oxygen as the sole oxidant have also been developed over the years [6]. Recently, to overcome the drawback from the use of toxic reagents or heavy metals, new metal-free oxidative coupling methods have been investigated. In 2005, Ochiai et al. first reported the iodobenzene-catalysed α-acyloxylation of ketones with acetic acid [7]. In 2011, Ishihara and co-workers reported the direct α-acyloxylation of carbonyl compounds with carboxylic acids under TBHP and TBAI conditions (Scheme 1a) [8]. Encouraged by this TBHP/TBAI system, chemists have achieved immense progress in the α-acyloxylation of ketones by using different substrates and mild oxidants, such as TBHP, K2S2O8 and H2O2 [9]. While each of these approaches represents an important advance towards the objective of a general method for the synthesis of α-acyloxycarbonyl compounds, each of them still utilizes organic oxidants to complete the C-O coupling process. Although Tomkinson and co-workers have reported the α-acyloxylation of carbonyl compounds by treating ketones with N-methyl-O-benzoylhydroxylamine hydrochloride (Scheme 1b) [10], this method still needs a three-step reaction process to synthesize the substrates. Therefore, the development of a facile, environmentally benign and efficient protocol for the preparation of α-acyloxycarbonyl compounds from simple and readily available starting materials with a broad substrate scope is still highly desirable.

|
Download:
|
| Scheme 1. Direct α-acyloxylation of ketones. | |
In this paper, we report the 1, 2-dibromoethane (EDB)-and potassium iodide (KI)-mediated α-acyloxylation of ketones with carboxylic acids. The most important features of the present catalytic system include: (1) the direct use of commercially available materials, (2) transition-metal-free catalytic systems and (3) milder reaction conditions without the use of strong oxidants, wide functional group compatibility and a broad substrate scope.
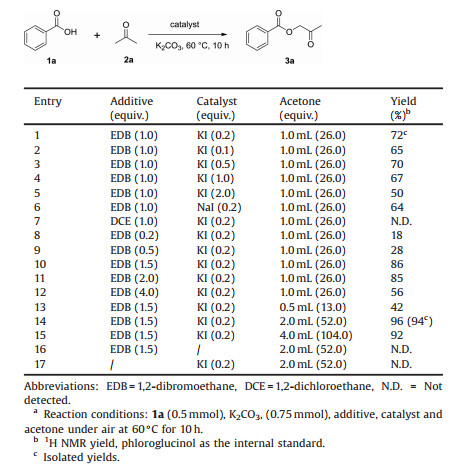
In the investigation of the reaction conditions (Table 1), benzoic acid 1a (1.0 equiv.) was initially treated with acetone 2a (1 mL) in the presence of EDB (1.0 equiv.), potassium iodide (KI, 0.2 equiv.), and potassium carbonate (K2CO3, 1.5 equiv.) at 60 ℃ for 10 h to afford 2-oxopropyl benzoate 3a, which was isolated by column chromatography in 72% yield (entry 1). The yield did not improve when examining different amounts of KI (entries 2-5). However, when we tried to replace the KI with NaI, the yield decreased to 64%, and 3a was not detected when EDB was substituted with 1, 2-dichloroethane (entries 6 and 7). Notably, we observed a satisfactory yield of 86% when the amount of additive EDB was increased to 1.5 equiv. (entry 10), and a further increase in EDB to 2.0 or 4.0 equiv. did not improve the yield of 3a (entries 11 and 12). Subsequently, we found that 2 mL of acetone 2a might be the most suitable equivalent, of which the isolated yield was improved to 94%, after evaluating a variety of different amounts of 2a in the reaction (entries 13-15). Finally, it is important to mention that product 3a was not detected in the absence of KI or EDB (entries 16 and 17). After a series of screening experiments, the optimal reaction conditions were obtained when 1a was treated with an excess of acetone 2a (2 mL, 52.0 equiv.), EDB (1.5 equiv.) and KI (0.2 equiv.) at 60 ℃.
|
|
Table 1 Optimization of the reaction conditions.a |
{kind=link}
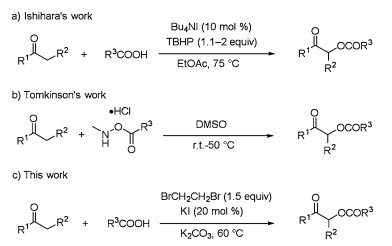
With the optimized reaction conditions in hand, we next evaluated the scope of the procedure by testing various carboxylic acids with acetone. First, a series of aromatic carboxylic acids were examined as shown in Scheme 2. Substrates with both electrondonating and electron-withdrawing substituents afforded the products in good to excellent yields under the standard conditions (3b-3p). Notably, hydroxy (3d and 3j), amino (3e) and aldehyde (3l) groups on the aromatic ring were well tolerated, thus offering opportunities for further derivatization. Heterocyclic aromatic carboxylic acids were also investigated and afforded the desired products in 53%-91% yield (3m-3p). To test whether this method could be extended to other carboxylic acids, we examined the reaction of aliphatic carboxylic acids with acetone. To our excitement, the desired products were obtained in 65%-94% yield (4a-4l). Notably, cinnamic acid, crotonic acid and propiolic acid provided the desired products in high yield (4a-4c). Further exploration of acyloxylation to aliphatic carboxylic acids also worked smoothly and afforded the coupling products in very good yields (4d-4h). Notably, the hydroxy and N-protected amino groups on the aliphatic chain were well tolerated (4i and 4j). It should be mentioned that steric hindrance proximal to the acid group also provided good yield, as exemplified by the successful synthesis of 4k and 4l (89% and 90% yield, respectively). We also attempted to use potassium phthalimide and 4-hydroxyacetophenone with acetone under the standard reaction conditions to afford the corresponding products 5a in 47% and 5b in 50% yield, respectively.

|
Download:
|
| Scheme 2. Scope of carboxylic acids. Reaction conditions: carboxylic acid (2.0 mmol), EDB (3.0 mmol), KI (0.4 mmol), K2CO3 (3.0 mmol), 8 mL acetone under air at 60 ℃. Isolated yields. | |
{kind=link}
To further investigate the utility of the EDB-and KI-mediated α-acyloxylation of a carboxylic acid with acetone in preparative organic synthesize, a gram-scale reaction was conducted. A total of 2.44 g of benzoic acid was treated with 40 mL of acetone 2a (26.0 equiv.) to yield 2.98 g of product 3a (84%), which was in accordance with the optimized reaction conditions (Table 1, entry 10).
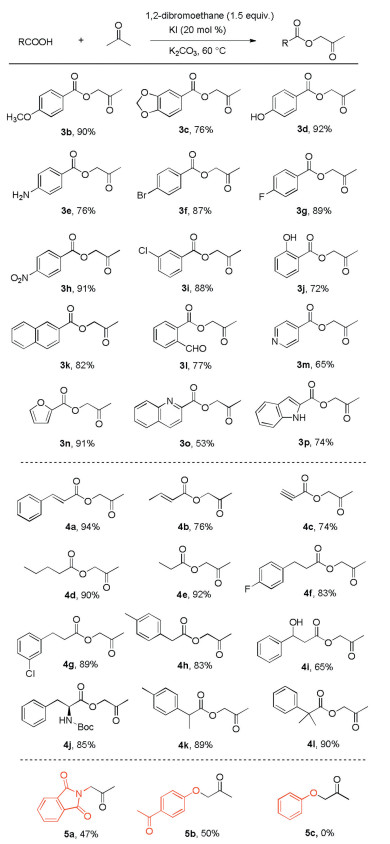
After demonstrating a series of acids, we then investigated the scope of ketones with benzoic acid. Considering the excess use of acetone under our optimized conditions, we attempted to reduce the amount of ketone and use DMF as the solvent, as shown in Scheme 3. We first treated benzoic acid with both an excess and 1.0 equivalent of cyclohexanone, which provided the target products in 62% and 32% yield respectively (6a). When 3-(3-chlorophenyl)propanoic acid and cinnamic acid were treated with excess cyclohexanone, the desired products were isolated in 59% and 53% yield, respectively (6b and 6c). An excess of 3-pentanone reacted with benzoic acid to provide the product in 45% yield (6d). We then turned our sights to the use of 1.0 equiv. of ketones. To our delight, acetophenone and 4-fluoroacetophenone reacted with benzoic acid to obtain desired products in 35% and 32% yield (6e and 6f), respectively. Different substituted substrates of propiophenone were examined with benzoic acid to obtain the desired products in good yield (6g-6i). 1-Phenylbutan-1-one also gave the target product in 35% yield (6j). However, it should be noted that 2-methyl-1-phenylpropan-1-one was not suitable for the transformation, which indicated that steric effects had a strong influence on the transformation. 2, 3-Dihydro-1H-inden-1-one and 5-fluoro-2, 3-dihydro-1H-inden-1-one were also tested to generate the products in 30% and 34% yield (6l and 6m), respectively.

|
Download:
|
| Scheme 3. Scope of ketones. Isolated yields. Reaction conditions: a ketone (5 mL), carboxylic acid (2.0 mmol), EDB (3.0 mmol), KI (0.4 mmol), K2CO3 (3.0 mmol) under air at 60 ℃; b ketone (2.0 mmol), carboxylic acid (3.0 mmol), EDB (3.0 mmol), KI (0.4 mmol), K2CO3 (3.0 mmol), 5 mL DMF under air at 60 ℃. | |
{kind=link}
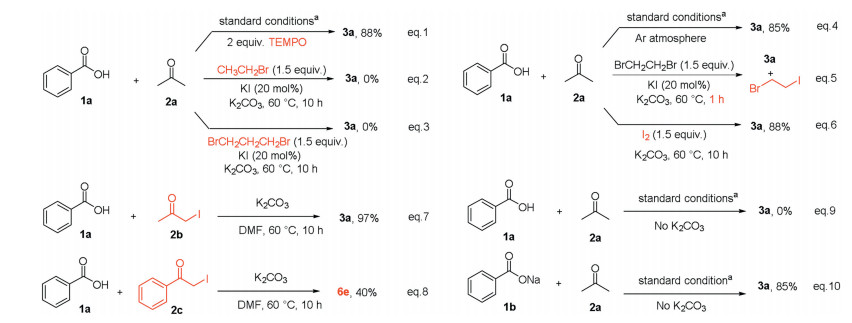
The following control experiments were carried out to gain insight into the mechanism of the reaction. First, target product 3a was isolated in 88% yield when 2 equiv. of 2, 2, 6, 6-tetramethylpiperidinyloxy (TEMPO) as a radical scavenger was added to the standard reactions, indicating that this transformation may not proceed via a radical pathway (Scheme 4, Eq. (1)). The generation of I2 was observed when treating EDB and KI in acetone at 60 ℃ while the I2 was not found in the absence of EDB (details in Supporting information). Besides, bromoethane and 1, 3-dibromopropane were used to replace EDB under the optimized conditions, which failed to provide the desired product (Scheme 4, Eqs. (2) and (3)). The reaction was also carried out under an argon atmosphere, which afforded product 3a in 85% yield (Scheme 4, Eq. (4)). The above experiments indicated that the EDB plays an important role in the generation of I2. In addition, the generation of 1-bromo-2-iodoethane was detected by GC-MS when reduced the reaction processtoone hour (Scheme 4, Eq. (5)). The product 3a also formed when we replaced EDB and KI with I2 (Scheme 4, Eq. (6)). Remarkably, the α-iodoacetone 2b and α-iodoacetopheneone 2c both could react with benzoic acid to afford the desired products 3a and 6e, respectively (Scheme 4, Eqs. (7) and (8)). Finally, the direct use of sodium benzoate 1b obtained 3a in 85% yield, while benzoic acid 1a failed to afford product 3a without the addition of K2CO3, which demonstrated that a carboxylic anion might be a key intermediate (Scheme 4, Eqs. (9) and (10)).

|
Download:
|
| Scheme 4. Control experiments to determine the reaction mechanism. | |
{kind=link}
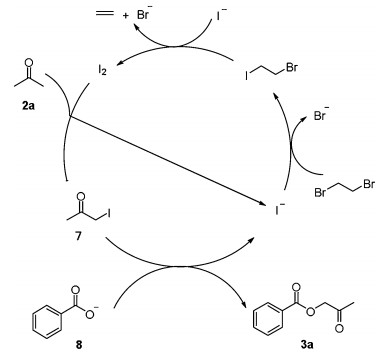
Based on the above control experiments and previous literature reports, a plausible mechanism was proposed in Scheme 5. First, the iodine anion reacted with 1, 2-dibromoethane to form 1-bromo-2-iodoethane, which then reacted with the iodine anion to give molecular iodine [11]. The subsequent reaction between iodine and acetone generated the intermediate 1-iodoacetone 7 [12]. Meanwhile, benzoic acid 2a was transformed into benzoate 8 in the presence of base. Finally, the reaction between intermediate 7 and benzoate 8 generated the desired product 3a.

|
Download:
|
| Scheme 5. Proposed reaction mechanism. | |
{kind=link}
In conclusion, we have developed the 1, 2-dibromoethane-and KI-mediated α-acyloxylation of ketones with carboxylic acids. Various acids, including aromatic and aliphatic acids, are well tolerated with wide functional group compatibility, and a series of ketones were also investigated. This protocol provides a convenient and environmentally benign method for the synthesis of α-acyloxycarbonyl compounds without the use of transition metal and strong oxidants, even on a gram scale. An 1, 2-dibromoethane and KI-catalysed reaction mechanism is proposed based on the results of control experiments. Further investigation of the relevant reactions is underway in our laboratory.
AcknowledgmentsWe are grateful to the Drug Innovation Major Project (No.2018ZX09711001-001-001) andCAMS Innovation Fund for Medical Sciences (CIFMS) (No. 2016-I2M-1-010) for financial support.
Appendix A. Supplementary dataSupplementary material related to this article can be found, in the online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.052.
| [1] |
(a) W. Huang, J. Pei, B. Chen, et al., Tetrahedron 52 (1996) 10131-10136; (b) M. Shindo, Y. Yoshimura, M. Hayashi, et al., Org. Lett. 9 (2007) 1963-1966; (c) S.P. Cutulic, N.J. Findlay, S.Z. Zhou, et al., J. Org. Chem. 74 (2009) 8713-8718; (d) R.N. Reddi, P.V. Malekar, A. Sudalai, Org. Biomol. Chem. 11 (2013) 6477-6482; (e) C. Lv, Q. Tu, J. Gong, et al., Tetrahedron 73 (2017) 3612-3621. |
| [2] |
(a) M. Fujita, T. Hiyama, J. Org. Chem. 53 (1988) 5415-5421; (b) M. Hojo, H. Harada, H. Ito, et al., J. Am. Chem. Soc. 119 (1997) 5459-5460; (c) M.R. Grimmett, Imidazoles, in: R. Neier (Ed.), Science of Synthesis, Georg Thieme Verlag, Stuttgart, 2002, pp. 325-528; (d) G. Scheid, W. Kuit, E. Ruijter, et al., Eur. J. Org. Chem. (2004) 1063-1074; (e) P.C. Patil, F.A. Luzzio, D.R. Demuth, Tetrahedron Lett. 56 (2015) 3039-3041. |
| [3] |
P.A. Levine, A. Walti, Organic Syntheses, Wiley & Sons, New York, 1943, pp. 5 Collect vol. 2.
|
| [4] |
T. Shinada, T. Kawakami, H. Sakai, et al., Tetrahedron Lett. 39 (1998) 3757-3760. DOI:10.1016/S0040-4039(98)00578-4 |
| [5] |
(a) For selectedexamples, see:, J.D. Cocker, H.B. Henbest, G.H. Phillipps, et al., J. Chem. Soc. (1965) 6-11; (b) M.E. Kuehne, T.J. Giacobbe, J. Org. Chem. 33 (1968) 3359-3369; (c) G.J. Williams, N.R. Hunter, Can. J. Chem. 54 (1976) 3830-3832; (d) T. Satoh, S. Motohashi, K. Yamakawa, Bull. Chem. Soc. Jpn. 59 (1986) 946-948; (e) J.C. Lee, Y.S. Jin, J.H. Choi, Chem. Commun. (2001) 956-957. |
| [6] |
(a) J. Du, X. Zhang, X. Suna, et al., Chem. Commun. 51 (2015) 4372-4375; (b) W.G. Jia, H. Zhang, D.D. Li, et al., RSC Adv. 6 (2016) 27590-27593; (c) X. Huang, X. Liang, J. Yuan, et al., Org. Chem. Front. 4 (2017) 163-169. |
| [7] |
M. Ochiai, Y. Takeuchi, T. Katayama, et al., J. Am. Chem. Soc. 127 (2005) 12244-12245. DOI:10.1021/ja0542800 |
| [8] |
M. Uyanik, D. Suzuki, T. Yasui, et al., Angew. Chem. Int. Ed. 50 (2011) 5331-5334. DOI:10.1002/anie.201101522 |
| [9] |
(a) S. Guo, J.T. Yu, Q. Dai, et al., Chem. Commun. 50 (2014) 6240-6242; (b) B. Mondal, S.C. Sahoo, S.C. Pan, Eur. J. Org. Chem. 14 (2015) 3135-3140; (c) Z. Zhou, J. Cheng, J.T. Yu, Org. Biomol. Chem. 13 (2015) 9751-9754; (d) L.M. Chang, G.Q. Yuan, Tetrahedron 72 (2016) 7003-7007; (e) C. Li, T. Jin, X. Zhang, et al., Org. Lett. 18 (2016) 1916-1919; (f) Y.D. Wu, B. Huang, Y.X. Zhang, et al., Org. Biomol. Chem. 14 (2016) 5936-5939; (g) C. Chen, W. Liu, P. Zhou, et al., RSC Adv. 7 (2017) 20394-20397; (h) P.S. Kumar, B. Ravikumar, K.C. Ashalu, et al., Tetrahedron Lett. 59 (2018) 33-37.0_TD$DIF] |
| [10] |
C.S. Beshara, H. Hall, R.L. Jenkins, et al., Org. Lett. 7 (2005) 5729[3-5732.
|
| [11] |
(a) C.F. van Duin, Rec. Trav. chim. 43 (1924) 341-358; (b) J. Hine, W.H. Brader, J. Am. Chem. Soc. 77 (1955) 361-364; (c) W.M. Schubert, H. Steadly, B.S. Rabinovitch, J. Am. Chem. Soc. 77 (1955) 5755. |
| [12] |
(a) R.P. Bell, K. Yates, J. Chem. Soc. (1962) 2285-2290; (b) E. Tapuhi, W.P. Jencks, J. Am. Chem. Soc. 104 (1982) 5758-5765; (c) M.L.N. Rao, D.N. Jadahav, Tetrahedron Lett. 47 (2006) 6883-6886. |