 2020, Vol. 31
2020, Vol. 31 


Quinolinone fused tetrahydropyran skeletons have received enormous attention as a ubiquitous structural unit in drugs and biologically active compounds [1]. Being an important constituent of quinolinones fused tetrahydropyrans family, tetrahydropyran [3, 2-c]quinolinones is also an important structural motif presented in plenty of natural products or synthetic compounds displaying a wide range of biological activities (Fig. 1) [2]. For example, natural product euodenine A was identified as an agonist of the human TLR4 receptor [3]. Zanthodioline was found to have strong antiplatelet aggregation activity in vitro [4]. Toddacoumalone isolated from Toddalia asiatica was reported as an effective phosphodiesterase-4 inhibitor with an IC50 around 140 nmol/L [5]. Despite the importance of those bioactive tetrahydropyran[3, 2-c]quinolinone molecules, the enantioselective synthesis of these kind of heterocycles are rarely reported. Therefore, the diverse biological profiles of these tetrahydropyran[3, 2-c]quinolinones prompted us to develop a novel and efficient catalytic method for the enantioselective synthesis of structurally diverse tetrahydropyran fused quinolinones.

|
Download:
|
| Fig. 1. Some bioactive pyrano[3, 2-c]quinolinones. | |
Construction of six-membered heterocycles with adjacent stereogenic centers has gain extensive research interests in recent years, especially by using the cycloaddition strategies. Among all the methods that have been developed, [3 + 3] annulation strategy was much less explored compared with [4 + 2] strategies [6]. Several asymmetric synthetic strategies to construct the chiral tetrahydropyran skeletons have been developed successfully in recent years [7]. Among all the strategies that have been reported, the organocatalyzed [3 + 3] annulation behaved as efficient methods [8]. For example, Gurubrahamam et al. successfully employed sequential organocatalyzed and DABCO-catalyzed olefin isomerization for asymmetric nucleophilic conjugate addition of hydroxycoumarins and pyranone to branched nitro enynes to afford pyrano-annulated scaffolds [9]. The similar strategy was used by Modrocká and Zhang et al. to construct pyrano[3, 2-c] chromen-5-on products [10]. Besides, Zheng et al. developed a bifunctional squaramide catalyzed enantioselective formal [3+3] annulation reactionwith pyrazolin-5-ones and nitroallylic acetates for the synthesis of tetrahydropyrano[2, 3-c]pyrazoles [11]. Despite few heterocycle fused tetrahydropyrans have been reported, most of them are oxygen-containing heterocycle and nitrogen-containing fused tetrahydropyrans are rarely. More importantly, nitrogencontaining heterocyclic compounds are more abundant in drugs and with great potential [12]. Thus, the construction of nitrogencontaining heterocycles fused tetrahydropyrans is of highly importance.
Based on these research above and previous work of our group, we observed that quinolinone 1 is a highly reactive nucleophile and could undergo nucleophilic addition twice (Scheme 1) [13]. Considering that Morita-Baylis-Hillman (MBH) esters of nitroalkenes 2, which commonly behaved as Michael acceptor, followed by a substitution reaction owing to the good leaving ability of the ester groups [14]. Thus, we hypothesized that chiral tetrahydropyrano[3, 2-c]quinolinones 3 could be conveniently constructed through an asymmetric Michael addition of 1, 3-binucleophilic quinolinones 1 to MBH esters of nitroalkenes 2, followed by the in situ intramolecular annulation reaction. The stereochemistry of this process could be controlled by chiral organo-catalyst. Herein, we reported a cascade two steps asymmetric organocatalytic Michael reaction from inter- to intra- stepwise to access tetrahydropyrano[3, 2-c]quinolinones in good yield, high diastereo- and enantioselectivities.

|
Download:
|
| Scheme 1. Construction of tetrahydropyrano[3, 2-c]quinolinone scaffolds via [3+3] annulation strategy. | |
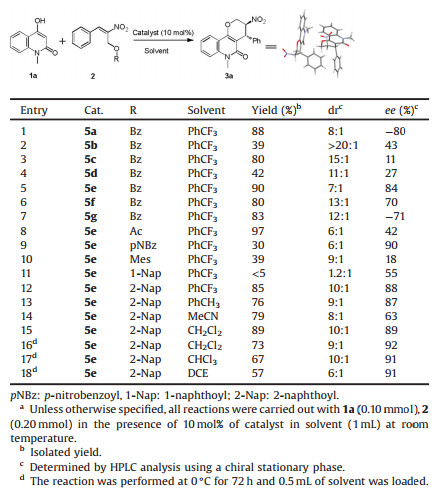
The research was initiated by testing the model reaction of easily prepared 4-hydroxy-1-methylquinolin-2(1H)-one (1a) [15] and (E)-2-nitro-3-phenylallyl benzoate (2a) [14a] in the presence of catalyst 5a (quinine) in PhCF3 (1 mL) at room temperature. To our delight, the reaction ran smoothly to afford the desired tetrahydropyran fused quinolinone derivative 3a in 88% yield with 8:1 dr and 80% ee (Table 1, entry 1). Several catalysts (Fig. 2), including H-bond donating bifunctional catalysts 5b and 5c, primary amine catalyst 5d and other natural cinchona alkaloids 5e, 5f and 5g were examined with the purpose of improving the stereoselectivity of this reaction (Table 1, entries 2–7). 5e (quinidine) turned out to be the best catalyst for this reaction, as it gave the reaction in 90% yield 7:1 dr and 84% ee (Table 1, entry 5). Perhaps not surprised, catalysts 5a and 5e gave the product 3a in the opposite configuration. Subsequently, we screened different protecting groups of MBH esters 2 under room temperature with the aim of further improving the stereoselectivity by using catalyst 5e (Table 1, entries 8–12). The reaction has good diastereo- and enantioselectivity with p-nitrobenzoyl as protecting group (6:1 dr, 90% ee), however low yield was observed (30%) (Table 1, entry 9). To our delight, a considerable improvement in both the yield (85%) and dr (10:1) was achieved by replacing the protecting group with 2-naphthoyl, despite the slightly diminished ee value (88%) (Table 1, entry 12). Other protecting groups like acetyl or 2, 4, 6- trimethylbenzoyl were either gave the reaction in poor enantioselectivity, or in low yield (Table S1 in Supporting information). Taken yield, dr and ee values into consideration, we choose 2- naphthoyl as the protecting group for further optimization. Next, solvents, reaction concentration, temperatures and time were screened by using catalyst 5e (Table 1, entries 13–18). Finally, the best reaction condition was established as performing the reaction in dichloromethane at 0 ℃ for 72 h, with the concentration of 0.2 mol/L, and gave the reaction in 73% yield, 9:1 dr and 92% ee (Table 1, entry 16). The molecular structure and absolute configuration of the two chiral centers within (3S, 4R)-(-)-3a were unambiguously determined by X-ray crystallography analysis (Table 1, Fig. S3 in Supporting information), and the crystallographic data has been deposited in cambridge centre with CCDC 1912415.
|
|
Table 1 Optimization of the reaction conditions.a |

|
Download:
|
| Fig. 2. Catalysts investigated in the reaction. | |
After establishing the optimized reaction conditions, we set out to explore the substrate scope of this asymmetric organocatalyzed reaction to provide the generality of this method by using catalyst 5e (Table 2). First, a variety of quinolinones 1 (Fig. S1 in Supporting information) were tested. The reaction worked well with quinolinones bearing different N-substituent groups (3b, 3c), both phenyl and benzyl substituted quinolinones gave the reaction in good results (Table 2, entries 1–2). When quinolinones have either electron-withdrawing substituents, or electron-donating substituents, or substituents on different positions on the benzene ring, they were well tolerated and delivered the corresponding cyclized products 3d–3f and 3g–3l in moderate to good yields (49%-95%) and high stereoselectivities (6:1 -> 20:1 dr, 87%-96% ee) (Table 2, entries 3–11). However, the reaction would not occur when the quinolinone without an N-substituent group, which probably due to this structure prone to form an aromatic system and reduce the nucleophilicity. We next investigated the versatility of MBH esters 2 (Fig. S2 in Supporting information) in this asymmetric reaction. We examined the electronic effect of the substituents on the benzene ring of the MBH esters 2. Generally, aromatic rings bearing electron-withdrawing or electron donating groups were turned out to be good candidates for the reaction, and gave the reaction in up to 95% yield and 95% ee (3m-3p, 3r-3t) (Table 2, entries 12–15, 17–19).
|
|
Table 2 Substrate scope of the asymmetic [3+3] annulation reaction.a |

{kind=link}
{kind=link}
{kind=link}
We noticed that substrates 2 with either meta- or parasubstituents on the benzene ring gave more satisfactory yields than those with ortho-substituents, probably due to the higher steric hindrance arised from the ortho-substituents. MBH esters 2 derived from 2-naphthyl nitroalkene turned out to be a suitable substrate and delivered the desired cyclic product 3q in high yield and good ee, whereas 2-furyl nitroalkene derived MBH esters only gave moderate yield and ee (Table 2, entries 16 and 20). Interestingly, (E)-styryl substituted MBH benzoate could react with quinolinone to give cyclic adduct 3v, albeit the low yield and moderate enantioselectivity (Table 2, entry 21). However, the reaction would not occur when MBH 2-naphthoates of aliphatic nitroolefins was used, which may due to the low reactivity of aliphatic nitroolefins. Moreover, other different heterocycles that contain the 1, 3-dicarbonyl structure are also applicable with similar results, gave 3w and 3x in good yield and high ee. Bicyclic product 3y was obtained in low yield, which might be attributable to the low nucleophilicity of the corresponding 1, 3-dicarbonyl compound 1o (Fig. S2).
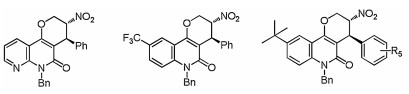
As we have observed that catalyst 5a gave the product in opposite configuration compared with 5e, we also briefly examined the substrate scope of the 5a catalyzed asymmetic [3 + 3] annulation reaction to further explore the generality of this asymmetric reaction. As shown in Fig. 3, this reaction exhibited good substrate compatibility. Quinolinone analog 1m reacted with MBH ester 2b to give 4a in good yield, excellent diastereoselectivity and enantioselectivitiy. Quinolinone bearing a trifluoromethyl or tert-butyl group on the aromatic ring also participate in the reaction well, and gave the corresponding adducts 4b–4e up to 91% yield and 95% ee (Fig. 3). Thus, by employing different catalysts, we could efficiently prepare a variety of chiral tetrahydropyrano[3, 2-c] quinolinones and their enantiomers.

|
Download:
|
| Fig. 3. Asymmetic [3 + 3] annulation reaction catalyzed by 5a under the optimized conditions. The yield is isolated yield. The ee value and dr are determined by HPLC analysis using a chiral stationary phase. | |
{kind=link}
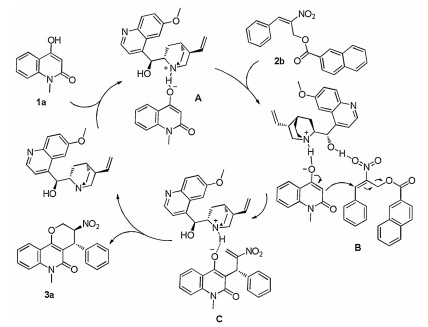
To illustrate the reaction mechanism conveniently, a plausible mechanism was proposed. As show in Scheme 2, intermediate A could be formed via a hydrogen-bonding interaction between the catalyst quinidine and substrate 1a. Then intermediate B was formed in a similar way and the reaction enantioselectivity was controlled in this process. Then inter-molecular Michael addition and the recovering of the catalyst were occur, which resulted in the formation of intermediate C. Finally, the desired annulation product 3a was formed via an intramolecular annulation reaction.

|
Download:
|
| Scheme 2. Proposed mechanism. | |
{kind=link}
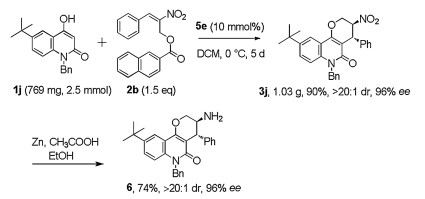
Finally, in order to demonstrate the potential practical utility of our method, we performed the reaction with 2.5 μmol of starting material 1j and reduced the equivalent of 2b to 1.5 equiv. By simply extended the reaction time, the reaction ran smoothly and delivered 1.03 g of tetrahydropyran[3, 2-c] quinolinone 3j with a 90% yield, > 20:1 dr and 96% ee. The result was almost identical to the reaction carried out in small scale, indicating that the reaction could be applied to prepare chiral building blocks in a large amount. In addition, the nitro group can easily transform into amino group under the mild reaction condition without any appreciable loss in the diastereo- and enantioselectivity, suggesting an additional potential utility of this chiral scaffold (Scheme 3).

|
Download:
|
| Scheme 3. Scale up reaction and transformation study. | |
{kind=link}
In summary, we developed an organocatalyzed asymmetric formal [3 + 3] annulation of 4-hydroxyquinoline-2(1H)-one and MBH 2-naphthoates of nitroolefins catalyzed by natural cinchona alkaloids. The reactions run smoothly to provide a variety of tetrahydropyrano[3, 2-c]quinolinones and analogs with good yields (up to 95%), high diastereo- and enantioselectivities (up to > 20:1 dr, 96% ee) under the optimized reaction condition. By employing different catalysts, the reactions could be performed in an enantioswitchable manner and gave both enantiomers in good yield and excellent enantioselectvities, respectively. In addition, this reaction could be carried out in a gram scale, which has potential practical utility. Using this synthetic strategy in preparing bioactive compounds are currently underway in our lab.
AcknowledgmentsWe are grateful for the financial support from the National Natural Science Foundation of China (No. 81602972), Guangdong Natural Science Funds for Distinguished Young Scholar (No.2018B030306017), Guangdong Province Universities and Colleges Pearl River Scholar Funded Scheme (2018).
Appendix A. Supplementary dataSupplementary material related to this articlecan be found, inthe online version, at doi:https://doi.org/10.1016/j.cclet.2019.08.040.
| [1] |
(a) T. Shiro, T. Fukaya, M. Tobe, Eur. J. Med. Chem. 97 (2015) 397-408; (b) B. Liu, F. Li, T. Zhou, X.Q. Tang, G.W. Hu, J. Heterocycl. Chem. 55 (2018) 1863-1873; (c) T. Yasumoto, Chem. Rec. 1 (2001) 228-242; (d) A. Bermejo, B. Figadère, M.C. Zafra-Polo, et al., Nat. Prod. Rep. 22 (2005) 269-303; (e) M. Murata, T. Yasumoto, Nat. Prod. Rep. 17 (2000) 293-314. |
| [2] |
(a) V. Nadaraj, S. Thamarai Selvi, H. Pricilla Bai, S. Mohan, T. Daniel Thangadurai, Med. Chem. Res. 21 (2011) 2902-2910; (b) Z.D. Yang, D.B. Zhang, J. Ren, M.J. Yang, Med. Chem. Res. 21 (2012) 722-725. |
| [3] |
J.E. Neve, H.P. Wijesekera, S. Duffy, et al., J. Med. Chem. 57 (2014) 1252-1275. DOI:10.1021/jm401321v |
| [4] |
I.S. Chen, I.W. Tsai, C.M. Teng, et al., Phytochemistry 46 (1997) 525-529. DOI:10.1016/S0031-9422(97)00280-X |
| [5] |
T.T. Lin, Y.Y. Huang, G.H. Tang, et al., J. Nat. Prod. 77 (2014) 955-962. DOI:10.1021/np401040d |
| [6] |
(a) X.X. Sun, H.H. Zhang, G.H. Li, Y.Y. He, F. Shi, Chem. -Eur. J. 22 (2016) 17526-17532; (b) Z.Q. Zhu, L. Yu, M. Sun, G.J. Mei, F. Shi, Adv. Synth. Catal. 360 (2018) 3109-3116; (c) X.X. Sun, C. Li, Y.Y. He, et al., Adv. Synth. Catal. 359 (2017) 2660-2670; (d) W. Dai, H. Lu, F. Shi, S.J. Tu, Chem. -Eur. J. 20 (2014) 11382-11389; (e) X. Yang, Y.C. Zhang, Q.N. Zhu, M.S. Tu, F. Shi, J. Org. Chem. 81 (2016) 5056-5065; (f) F. Shi, R.Y. Zhu, W. Dai, C.S. Wang, S.J. Tu, Chem. -Eur. J. 20 (2014) 2597-2604. |
| [7] |
(a) A. Temperini, A. Barattucci, P.M. Bonaccorsi, O. Rosati, L. Minuti, J. Org. Chem. 80 (2015) 8102-8112; (b) C. Bosset, P. Angibaud, I. Stanfield, et al., J. Org. Chem. 80 (2015) 12509-12525; (c) T.J. Potter, J.A. Ellman, Org. Lett. 18 (2016) 3838-3841; (d) J. Scoccia, S.J. Pérez, V. Sinka, et al., Org. Lett. 19 (2017) 4834-4837; (e) Y. Kurimoto, T. Nasu, Y. Fujii, K. Asano, S. Matsubara, Org. Lett. 21 (2019) 2156-2160; (f) X. Xie, C. Peng, G. He, et al., Chem. Commun. 48 (2012) 10487-10489. |
| [8] |
(a) J.Y. Liu, X.C. Yang, H. Lu, Y.C. Gu, P.F. Xu, Org. Lett. 20 (2018) 2190-2194; (b) D.K. Nair, R.F.S. Menna-Barreto, E.N. da Silva Júnior, S.M. Mobin, I.N.N. Namboothiri, Chem. Commun. 50 (2014) 6973-6976; (c) R. Chen, X. Fan, J. Gong, Z. He, Asian J. Org. Chem. 3 (2014) 877-885; (d) C.Y. Yu, M. Yaqub, Y.M. Jia, Z.T. Huang, Synlett 9 (2008) 1357-1360; (e) W. Xiao, X. Yin, Z. Zhou, W. Du, Y.C. Chen, Org. Lett. 18 (2015) 116-119; (f) S. Chandrasekhar, K. Mallikarjun, G. Pavankumarreddy, K.V. Rao, B. Jagadeesh, Chem. Commun. (2009) 4985-4987; (g) B. Han, Y.C. Xiao, Z.Q. He, Y.C. Chen, Org. Lett. 11 (2009) 4660-4663; (h) M.L. Shi, G. Zhan, S.L. Zhou, W. Du, Y.C. Chen, Org. Lett. 18 (2016) 6480-6483; (i) L. Yang, W. Huang, X.H. He, et al., Adv. Synth. Catal. 358 (2016) 2970-2975. |
| [9] |
R. Gurubrahamam, B.F. Gao, Y.M. Chen, et al., Org. Lett. 18 (2016) 3098-3101. DOI:10.1021/acs.orglett.6b01265 |
| [10] |
(a) V. Modrocká, E. Veverková, M. Meciarová, R. Šebesta, J. Org. Chem. 83 (2018) 13111-13120; (b) J. Zhang, G. Yin, Y. Du, et al., J. Org. Chem. 82 (2017) 13594-13601. |
| [11] |
Y. Zheng, L. Cui, Y. Wang, Z. Zhou, J. Org. Chem. 81 (2016) 4340-4346. DOI:10.1021/acs.joc.6b00196 |
| [12] |
E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257-10274. DOI:10.1021/jm501100b |
| [13] |
(a) V. Nair, P.M. Treesa, C.N. Jayan, et al., Tetrahedron 57 (2001) 7711-7717; (b) A. Klásek, O. Rudolf, M. Rouchal, A. Lycka, A. Ru 9žicka, Tetrahedron 69 (2013) 9492-499; (c) Y. Gu, J. Barrault, F. Jérôme, Adv. Synth. Catal. 351 (2009) 3269-3278; (d) M. Rueping, E. Merino, M. Bolte, Org. Biomol. Chem. 10 (2012) 6201-6210; (e) Y.R. Lee, B.S. Kim, H.I. Kweon, Tetrahedron 56 (2000) 3867-3874; (f) Y.R. Lee, H.I. Kweon, W.S. Koh, et al., Synthesis (Stuttgart) (2001) 1851-1855; (g) X. Zhu, A. Lin, Y. Shi, et al., Org. Lett. 13 (2011) 4382-4385. |
| [14] |
(a) W. Xiao, X. Yin, Z. Zhou, W. Du, Y.C. Chen, Org. Lett. 18 (2016) 116-119; (b) L.F. Yeh, S. Anwar, K. Chen, Tetrahedron 68 (2012) 7317-7321; (c) D.K. Nair, S.M. Mobin, I.N.N. Namboothiri, Tetrahedron Lett. 53 (2012) 3349-3352. |
| [15] |
A. Dey, A. Hajra, Org. Biomol. Chem. 15 (2017) 8084-8090. DOI:10.1039/C7OB02124K |